

In this issue of Blood, identified novel pathogenetic missense mutations in the IKAROS family zinc finger 5 (IKZF5) gene that were associated with isolated thrombocytopenia in 3 pedigrees, affecting a total of 20 individuals and representing de novo mutations also associated with isolated thrombocytopenia in 3 individuals from 2 pedigrees.1
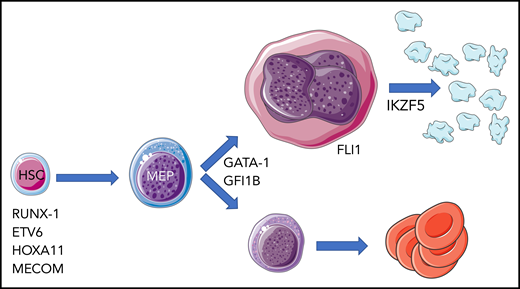
Hereditary thrombocytopenia associated with defects in transcription factor genes. The cellular level at which abnormal activity of mutated transcription factor is exerted is largely inferred by the associated hematologic abnormalities. HSC, hematopoietic stem cell; MEP, megakaryocyte-erythroid progenitor.
Hereditary thrombocytopenia associated with defects in transcription factor genes. The cellular level at which abnormal activity of mutated transcription factor is exerted is largely inferred by the associated hematologic abnormalities. HSC, hematopoietic stem cell; MEP, megakaryocyte-erythroid progenitor.
IKZF5 (also known as Pegasus [a winged horse in Greek mythology]) is one of the zinc finger transcription factors encoded by genes belonging to the IKAROS family that includes genes named after 4 additional mythological divinities: the founding member IKZF1 (Ikaros), IKZF2 (Helios), IKZF3 (Aiolos), and IKZF4 (Eos).2 The best known IKAROS family members (ie, IKZF1, -2, and -3) are involved in lymphocyte development, and somatic mutations of IKZF1 and IKZF3 are associated with hematologic malignances.3 Conversely, the functions of IKZF4 and IKZF5 remained elusive until the Lentaigne et al study established a role for IKZF5 in megakaryocytopoiesis.
Hereditary thrombocytopenias (HTs) are rare and presumably underdiagnosed diseases if, according to an Italian study, their prevalence is estimated at almost 3 of every 100 000 individuals.4 HT should be suspected in the presence of an evocative familial history, concomitant congenital defects, if present, and/or the detection of morphologic and/or volumetric and/or functional platelet abnormalities. Assessment of these platelet abnormalities requires considerable expertise and complex laboratory tests, and these platelet abnormalities are subjected to high phenotypic variability. Major advances in the understanding of HTs came from the application of whole-exome sequencing or whole-genome sequencing (WGS) to phenotypically well-characterized kindreds. This approach has identified an unexpectedly high number of involved genes.5 This knowledge is promoting a shift in the classification of HTs from schemes based on associated (or not) syndromic features, the size and volume and/or functional defects of platelets, or the main pathophysiologic mechanisms responsible for thrombocytopenia to a genetic defect-based classification. However, it is estimated that only about 50% of HTs can currently be associated with defined genetic variants. It is noteworthy that a joint effort of the American Society of Hematology and the National Institutes of Health–funded ClinGen resource (http://clinicalgenome.org) for HT variant classification is underway. Accurate diagnosis of HTs, notwithstanding how infrequent they are, is clinically relevant because some patients may be long misdiagnosed as having autoimmune thrombocytopenia, which leads to inappropriate exposure to prolonged cortisone therapy, use of thrombopoietin mimetics, and/or splenectomy.6 Adopting the phenotypic criteria commonly used in clinical practice, thrombocytopenia associated with IKZF5 mutations might be classified as an isolated mild thrombocytopenia without additional clinical features, with normally sized platelets and modest, variable platelet functional abnormalities, likely the result of a defect in the very late stages of megakaryocytopoiesis.
There are several lessons that can be learned from the work of Lentaigne et al. The first is that IKZF5 mutations must be added to the list of autosomal (HOXA11, RUNX1, FLI1, GFI1B, ETV6, MECOM) and X-linked (GATA1) transcription factor–encoding genes whose germ line variants are associated with HT (see figure). Of importance, IKZF5-associated thrombocytopenia seems to be minimally symptomatic, with mild if any bleeding tendency, and to have an overall favorable outcome. This is quite different from other forms of HTs that are the result of transcription factor variants, in particular, the leukemia-predisposing diseases associated with RUNX1 and ETV67 mutations that pose clinical and ethical issues regarding the identification, counseling, and management of affected individuals.
The second is that the newly discovered mutations in IKZF5 are rare genetic determinants of HT; indeed, only 5 variants were identified among the 233 patients with isolated thrombocytopenia in the WGS data from 13 037 individuals enrolled in the National Institute for Health Research BioResource. Although the study by Lentaigne et al has the value of describing a novel molecularly characterized entity of HT, it leaves largely unchanged the number of patients with HT still in search of a genetic explanation for their platelet disorder.
Third, the functional analysis of mutated IKZF5 performed in their study establishes Pegasus as a novel transcriptional regulator of thrombocytopoiesis and opens avenues for further research. Detailed studies of gene expression profiles by RNA sequencing in platelets of IKFZ5-mutated individuals highlighted that almost 10% of the >10 000 expressed RNAs were abnormally regulated compared with just 4 RNAs in purified CD4+ T cells, monocytes, and neutrophils, which makes a strong case for IKZF5 being highly specific for the megakaryocytic lineage, in spite of being widely expressed in hematopoietic cells. Therefore, IKZF5-mutated megakaryocytes might represent a powerful model for elucidating mechanisms of platelet granule formation and platelet release via proplatelet formation. On the clinical side, this lineage specificity of action explains isolated thrombocytopenia as the only hematologic abnormality in patients with an IKZF5 mutation, at odds with patients who have mutations in GATA1 and GLI1B, who also present with anemia of various severities, or those with HOXA11 and MECOM abnormalities, in which thrombocytopenia is part of a more generalized hematopoietic failure. Thrombocytopenia seems to be the only hematologic abnormality that also appears in the Paris-Trousseau syndrome as a result of hemizygous FLI1 deletion, which is associated with the additional manifestations of the Jacobsen syndrome, or homozygous missense mutation leading to isolated thrombocytopenia (see figure).
Finally, the Lentaigne et al study represents an illuminating example of the incredible amount of information that population genome sequencing efforts have the potential to generate for the understanding of rare diseases, provided the right questions are asked and the right approach to interpretation of genetic data is implemented.
Conflict-of-interest disclosure: The author declares no competing financial interests.
