

Abstract
A hallmark of acute myeloid leukemia (AML) is epigenetic dysregulation, which is initiated by recurrent translocations and/or mutations in transcription factors and chromatin regulators. This manifests as a block in myeloid differentiation and an increase in malignant self-renewal. These common features of AML have led to widespread optimism that epigenetic therapies would dramatically change the natural history of this disease. Although preclinical studies with these drugs fueled this optimism, results from early clinical trials have offered a more sobering message. Here, we provide an overview of epigenetic therapies that are currently approved by therapeutic regulatory authorities across the world and those undergoing early-phase clinical trials. We also discuss the conceptual and molecular factors that may explain some of the disparity between the bench and bedside, as well as emerging avenues for combining the current generation of epigenetic therapies with other classes of agents and the development of novel epigenetic therapies. With further research and development of this exciting class of drugs, we may finally be able to dramatically improve outcomes for patients afflicted with this aggressive and often incurable malignancy.
Introduction
Acute myeloid leukemia (AML) is an aggressive hematological cancer that is characterized by malignant self-renewal and a block in myeloid differentiation. As a consequence of its poor prognosis and the ease with which primary malignant cells can be accessed, AML has been one of the most comprehensively studied cancers. Collaborative efforts from a large number of investigators aimed at annotating the genetic landscape have provided a near complete list of the somatic mutations and chromosomal translocations that initiate and maintain the disease.1-3 One of the most striking findings from these studies was that ∼70% of recurring mutations in AML patients target regulators of gene expression, including epigenetic proteins, transcription factors (TFs), and the components of the splicing machinery.1,2,4,5 This observation, together with the low mutational burden of AML, suggests that dysregulation of the epigenome likely has a prominent role in this particular malignancy.2
Overview of epigenetics
Broadly speaking, the term epigenetics has been used to describe the study of events that regulate access to the DNA template to facilitate transcription, DNA repair, and DNA replication. In every nucleated cell in our body, our genomes are packaged as chromatin, a macromolecular complex of histone proteins and DNA. There are 2 major higher-order chromatin structures that exist within each cell: the loosely packed euchromatin and the more condensely packed heterochromatin. Euchromatin provides an open and accessible chromatin landscape within which the expressed genes for a particular cell are located. Genes that are not expressed and the pericentromeric and peritelomeric regions of chromosomes are more tightly packaged into a relatively inaccessible and transcriptionally silent heterochromatic state that is mainly located at the nuclear periphery (Figure 1).
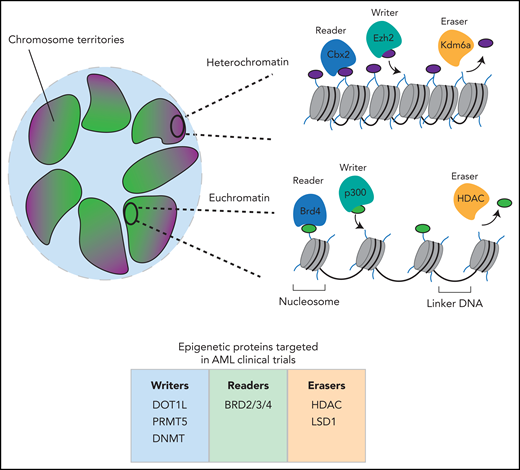
Overview of chromatin organization. Inside the nucleus of a cell, chromosomes exist in discrete chromosomal territories. The chromatin that makes up the chromosomes is packaged differently at different loci. Regions of euchromatin within these territories tend to cluster in the center of the nucleus, whereas regions of heterochromatin tend to associate with the periphery. In euchromatin, nucleosomes are less tightly packed and display histone modifications associated with transcriptional activation. In heterochromatin, the nucleosomes are tightly packed and have histone modifications associated with repression. Both active and repressive histone modifications are regulated by writer, reader and eraser proteins. Illustrated beneath the image is a summary of the currently used clinical compounds in AML that target epigenetic writers, readers, and erasers. BRD, bromodomain-containing protein; Cbx2, chromobox 2; DNMT, DNA methyltransferase; DOT1L, disruptor of telomeric silencing 1-like; Ezh2, enhancer of zeste homolog 2; HDAC, histone deacetylase; Kdm6A, lysine demethylase 6A; LSD1, lysine-specific histone demethylase 1; PRMT5, protein arginine N-methyltransferase 5.
Overview of chromatin organization. Inside the nucleus of a cell, chromosomes exist in discrete chromosomal territories. The chromatin that makes up the chromosomes is packaged differently at different loci. Regions of euchromatin within these territories tend to cluster in the center of the nucleus, whereas regions of heterochromatin tend to associate with the periphery. In euchromatin, nucleosomes are less tightly packed and display histone modifications associated with transcriptional activation. In heterochromatin, the nucleosomes are tightly packed and have histone modifications associated with repression. Both active and repressive histone modifications are regulated by writer, reader and eraser proteins. Illustrated beneath the image is a summary of the currently used clinical compounds in AML that target epigenetic writers, readers, and erasers. BRD, bromodomain-containing protein; Cbx2, chromobox 2; DNMT, DNA methyltransferase; DOT1L, disruptor of telomeric silencing 1-like; Ezh2, enhancer of zeste homolog 2; HDAC, histone deacetylase; Kdm6A, lysine demethylase 6A; LSD1, lysine-specific histone demethylase 1; PRMT5, protein arginine N-methyltransferase 5.
The fundamental repetitive unit of chromatin is called the nucleosome, which is made up of a histone octamer (containing 2 each of the histone proteins H2A, H2B, H3, and H4) around which 147 base pairs of DNA are wrapped. Decades of research have established that all components of the nucleosome (histone proteins and DNA) are chemically modified. These highly conserved histone and DNA modifications are specifically deposited by enzymes that are broadly termed “epigenetic writers,” which include histone methyltransferases such as enhancer of zeste homolog 2 (EZH2) and DNA methyltransferases such as DNA methyltransferase 3α (DNMT3A). Chromatin modifications are highly dynamic and are readily removed by the enzymatic action of a different class of proteins which are broadly termed “epigenetic erasers,” which include the histone deacetylase (HDAC) families that remove lysine acetylation on histones and the ten-eleven translocation (TET) family of enzymes that facilitate DNA demethylation. Chromatin modification generally serves 2 primary purposes. Some modifications alter the electrostatic interactions between DNA and histones, enabling the switching between euchromatic and heterochromatic states. Importantly, virtually all known modifications on histones and DNA also serve as molecular beacons for the binding of proteins that have specialized domains that recognize these modifications. This class of proteins are broadly termed “epigenetic readers” and include proteins such as bromodomain-containing protein 4 (BRD4) that bind acetylated lysines on histones via their bromodomain. It is also important to note that 2 adjoining nucleosomes are separated by a short stretch of DNA (∼50 bp) called linker DNA. Linker DNA is bound by specialized proteins that can facilitate DNA packaging or, in euchromatic regions that are transcriptionally active, it can be bound by TFs and other components of the transcriptional machinery that potentiate gene expression (Figure 1).
Due to the critical role of epigenetic proteins in regulating the balance between self-renewal and differentiation, it is unsurprising that many of the mutations found within AML genomes are located in epigenetic regulators. This observation has fueled significant interest from academic scientists, clinicians, and the pharmaceutical industry to develop, characterize, and trial small molecules that exploit the inherent plasticity of the epigenome and abrogate malignant transcription programs.6 Herein, we provide a perspective on the current landscape of epigenetic therapies in AML and what the future may hold for this promising class of drugs.
Current epigenetic therapies in AML
After nearly 4 decades of stagnation, the last few years have witnessed a dramatic change in the therapeutic landscape of AML. Across the world, new therapies have been approved for all patient demographics including newly diagnosed patients receiving intensive chemotherapy, patients with relapsed and refractory disease, elderly patients unable to tolerate intensive therapy, and patients with secondary AML. Remarkably, the newly approved agents span a broad class of therapies including kinase inhibitors, antibody-based therapies, inhibitors of apoptosis, and epigenetic therapies. An overview of the new therapeutic landscape in AML is shown in Figure 2. Here, we will focus on the approved and emerging epigenetic therapies.
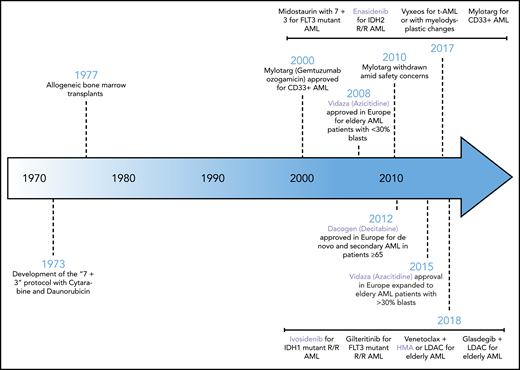
The history of approved therapies in AML. Shown here is a chronological illustration of therapies that have been approved by global therapeutic authorities for the management of AML. Highlighted in lavender are the epigenetic therapies. HMA, hypomethylating agent; IDH2, isocitrate dehydrogenase 2; LDAC, low-dose cytarabine; R/R, relapsed/refractory; t-AML, therapy-related AML.
The history of approved therapies in AML. Shown here is a chronological illustration of therapies that have been approved by global therapeutic authorities for the management of AML. Highlighted in lavender are the epigenetic therapies. HMA, hypomethylating agent; IDH2, isocitrate dehydrogenase 2; LDAC, low-dose cytarabine; R/R, relapsed/refractory; t-AML, therapy-related AML.
Epigenetic therapies approved by regulatory authorities across the world
DNA-hypomethylating agents
Much like the mutational architecture of AML cells, DNA methylation signatures can also segregate patients into distinct prognostic categories.7-9 Certain patterns in DNA methylation can be attributed to frequently recurring mutations in epigenetic regulators, such as those in DNMT3A, TET2, and isocitrate dehydrogenase 1/2 (IDH1/2) genes, which perturb the function of the main protagonists that catalyze the modification of cytosines.10,11 Mutations in DNMT3A are generally associated with global hypomethylation, whereas mutations in TET2 and IDH1/2 result in DNA hypermethylation.10,12 This suggests that the imbalance of DNA methylation in either direction is capable of supporting AML transformation, motivating attempts to pharmacologically reverse these oncogenic changes. The principal class of drugs used as DNA-hypomethylating agents are the nucleoside analogs. Azacitidine and decitabine freely incorporate into DNA (and RNA in the case of azacitidine) in replicating cells where they irreversibly bind and deplete the DNMTs, culminating in global hypomethylation.13,14 The incorporation of these drugs into DNA can lead to DNA damage and although the effects on DNA hypomethylation have been exhaustively studied, the concurrent contribution of the DNA-damage response to the clinical efficacy of these drugs remains largely unknown.15 These drugs have been widely assessed in clinical studies and were the first class of epigenetic drugs to be approved internationally for the management of high-grade myelodysplastic syndrome and AML with a blast count <30%. More recently, they have also been approved in combination with venetoclax for elderly AML patients who are not candidates for high-intensity chemotherapy.16-19 Interestingly, emerging evidence from clinical trials that have explored the use of hypomethylating agents as maintenance therapies have also demonstrated that azacitidine maintenance increased the time to relapse in older patients who obtained a complete remission following intensive chemotherapy.20
Although these agents have been widely used for decades, their pleiotropic effects have meant that elucidating their precise mechanism of action has been difficult. Without this understanding, improvement to their clinical efficacy has been limited. The primary mechanism by which hypomethylating agents were thought to exert their efficacy was by reactivation of tumor-suppressor genes that had been silenced by aberrant DNA methylation.21 Consequently, it was hoped that patients with mutations that induce DNA hypermethylation would have a greater response to hypomethylating agents. Although there is some evidence that patients with TET2 mutations derive greater benefit from these drugs, these findings are not seen in all patients with global hypermethylation, including those with IDH1/2 mutations.22,23 It is possible that clonal heterogeneity may confound the correlation between response and mutational status, however, it is also becoming increasingly apparent that these agents can trigger additional tumor-intrinsic or -extrinsic changes that contribute toward their efficacy.
A large body of emerging evidence suggests that DNA-hypomethylating agents can have direct and indirect immunomodulatory effects on malignant cells. They are able to enhance antitumor immunity in AML through derepression of several immune-related genes including cancer testis antigens and genes stimulated by interferon signaling pathways, including major histocompatibility complex class I.24,25 Additionally, these drugs have been shown to reactivate endogenous retroviruses (ERVs), triggering viral defense pathways and leading to upregulation of apoptotic and interferon response genes (Figure 3).26,27 As these drugs are given systemically, hypomethylating agents can also influence cells within the tumor microenvironment leading to indirect immunomodulatory effects.28 As a consequence of these exciting new insights, several clinical trials have recently commenced that combine hypomethylating agents with various immunotherapies to potentially further improve clinical outcomes (Table 1).25
![The effect of epigenetic therapies on cell-intrinsic modulation of the immune system. Epigenetic therapies including hypomethylating agents and inhibitors of LSD1, EZH2, and HDACs have been shown to reactivate epigenetically silenced endogenous retroviruses (ERVs). Removal of repressive marks by these epigenetic drugs results in transcription of ERVs, leading to the formation of double-strand RNA (dsRNA) molecules in the nucleus. These are then detected by pattern recognition receptors (eg, melanoma differentiation–associated protein 5 [MDA5]) in the cytoplasm that typically sense dsRNAs originating from viruses. This dsRNA sensing leads to a state of “viral mimicry” via the initiation of a signaling cascade mediated by mitochondrial antiviral signaling protein (MAVS) and results in upregulation of the TFs IFN regulatory factor 3/7 (IRF3/7) and NFKB to induce an interferon (IFN) type I/III response. IFNI/III signals in an autocrine manner through the interferon α receptor (IFNAR) to culminate in transcription of the IFN response genes. This ultimately leads to upregulation of key immune molecules such as major histocompatibility complex (MHC) class 1 on the cell surface. Bromodomain and extra-terminal motif (BET) inhibition has been associated with decreased expression of the inhibitory molecule programmed death ligand 1 (PD-L1), increasing the visibility of the leukemic cell to the immune system. Conversely, hypomethylating agents can lead to increased expression of PD-L1, rendering the cell sensitive to checkpoint inhibition. BETi, BET inhibitor; EZH2i, EZH2 inhibitor; HDACi, HDAC inhibitor; LSD1i, LSD1 inhibitor.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/134/22/10.1182_blood.2019003262/3/m_bloodbld2019003262f3.png?Expires=1769151603&Signature=Mwe5Y-T7dkPettM21YI-8IHV0FmYxcFq3ZsKCyjq5YGu5Qy94OulBXBIyXBepvTcRT0rpx0jKt~Xv9FOpGUn9mDfVYX~mxY0S4jdp9WGqur8ivRNPgOZEOccj~XEhHWZrc2LacZE91Q6FXcP~Bpn9yH7sjguhpS28CDS9DZ0cm-nrQbWwZuEJqgzGa-X45cQe5BQB5058BgzLJlWF9UVckGsFeBtpo7JP6g12z9KFlNVvZgCeG~ynY88r5PGKtSB6sdHXxmzCuxjYe5Oo5qkGTqGbgU0Z7EmgR3xkWM5EqfR4t88dESHZ8Ut~DBCMzJLXtqLpX5j9-gSj6rhapmg2Q__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
The effect of epigenetic therapies on cell-intrinsic modulation of the immune system. Epigenetic therapies including hypomethylating agents and inhibitors of LSD1, EZH2, and HDACs have been shown to reactivate epigenetically silenced endogenous retroviruses (ERVs). Removal of repressive marks by these epigenetic drugs results in transcription of ERVs, leading to the formation of double-strand RNA (dsRNA) molecules in the nucleus. These are then detected by pattern recognition receptors (eg, melanoma differentiation–associated protein 5 [MDA5]) in the cytoplasm that typically sense dsRNAs originating from viruses. This dsRNA sensing leads to a state of “viral mimicry” via the initiation of a signaling cascade mediated by mitochondrial antiviral signaling protein (MAVS) and results in upregulation of the TFs IFN regulatory factor 3/7 (IRF3/7) and NFKB to induce an interferon (IFN) type I/III response. IFNI/III signals in an autocrine manner through the interferon α receptor (IFNAR) to culminate in transcription of the IFN response genes. This ultimately leads to upregulation of key immune molecules such as major histocompatibility complex (MHC) class 1 on the cell surface. Bromodomain and extra-terminal motif (BET) inhibition has been associated with decreased expression of the inhibitory molecule programmed death ligand 1 (PD-L1), increasing the visibility of the leukemic cell to the immune system. Conversely, hypomethylating agents can lead to increased expression of PD-L1, rendering the cell sensitive to checkpoint inhibition. BETi, BET inhibitor; EZH2i, EZH2 inhibitor; HDACi, HDAC inhibitor; LSD1i, LSD1 inhibitor.
The effect of epigenetic therapies on cell-intrinsic modulation of the immune system. Epigenetic therapies including hypomethylating agents and inhibitors of LSD1, EZH2, and HDACs have been shown to reactivate epigenetically silenced endogenous retroviruses (ERVs). Removal of repressive marks by these epigenetic drugs results in transcription of ERVs, leading to the formation of double-strand RNA (dsRNA) molecules in the nucleus. These are then detected by pattern recognition receptors (eg, melanoma differentiation–associated protein 5 [MDA5]) in the cytoplasm that typically sense dsRNAs originating from viruses. This dsRNA sensing leads to a state of “viral mimicry” via the initiation of a signaling cascade mediated by mitochondrial antiviral signaling protein (MAVS) and results in upregulation of the TFs IFN regulatory factor 3/7 (IRF3/7) and NFKB to induce an interferon (IFN) type I/III response. IFNI/III signals in an autocrine manner through the interferon α receptor (IFNAR) to culminate in transcription of the IFN response genes. This ultimately leads to upregulation of key immune molecules such as major histocompatibility complex (MHC) class 1 on the cell surface. Bromodomain and extra-terminal motif (BET) inhibition has been associated with decreased expression of the inhibitory molecule programmed death ligand 1 (PD-L1), increasing the visibility of the leukemic cell to the immune system. Conversely, hypomethylating agents can lead to increased expression of PD-L1, rendering the cell sensitive to checkpoint inhibition. BETi, BET inhibitor; EZH2i, EZH2 inhibitor; HDACi, HDAC inhibitor; LSD1i, LSD1 inhibitor.
Combination strategies involving epigenetic therapies currently in clinical trials
| Drug | Targets | Phase | Date | Clinical trial |
|---|---|---|---|---|
| Combinations of epigenetic therapies | ||||
| Azacitidine + entinostat (MS275) | DNMT + HDAC | 2 | 2011 | NCT01305499 |
| Azacitidine + FT-2102 | DNMT1 + IDH1 | 1/2 | 2016 | NCT02719574 |
| Azacitidine + LSD1 inhibitor (NCB059872) | DNMT1 + LSD1 | 1/2 | 2016 | NCT02712905 |
| Azacitidine + ivosidenib (AG-120) | DNMT + IDH1 | 3 | 2017 | NCT03173248 |
| Azacitidine + pracinostat (SB939) | DNMT + HDAC | 3 | 2017 | NCT03151408 |
| Decitabine + vorinostat before or during FLAG | DNMT + HDAC | 1 | 2017 | NCT03263936 |
| Azacitidine + PRMT5 inhibitor (GSK3326595) | DNMT + PRMT5 | 1 | 2018 | NCT03614728 |
| Low-dose azacitidine + vorinostat after alloHSCT | DNMT + HDAC | 1 | 2019 | NCT03843528 |
| Epigenetic therapies in combination with immunotherapies | ||||
| Azacitidine + nivolumab (MDX-1106) +/or ipilimumab (MDX-010) | DNMT + PD-1 + CTLA-4 | 2 | 2015 | NCT02397720 |
| Azacitidine + pembrolizumab (MK-3475) | DNMT + PD-1 | 2 | 2016 | NCT02845297 |
| Decitabine + ipilimumab (MDX-010) | DNMT + CTLA-4 | 1 | 2016 | NCT02890329 |
| Guadecitabine + atezolizumab (MPDL 3280A) | DNMT + PD-L1 | 1/2 | 2016 | NCT02935361 |
| Decitabine + PDR001 +/or MBG453 | DNMT + PD-1 + TIM-3 | 1 | 2017 | NCT03066648 |
| Azacitidine + Hu5F9-G4 | DNMT + CD47 | 1 | 2017 | NCT03248479 |
| Decitabine + CDX-1401 + poly ICLC + nivolumab (MDX-1106) | DNMT + DEC-205 + TLR-3 + PD-1 | 1 | 2017 | NCT03358719 |
| Decitabine + avelumab | DNMT + PD-L1 | 1 | 2018 | NCT03395873 |
| Azacitidine + nivolumab (ADVL1412) | DNMT + PD-1 | 1/2 | 2019 | NCT03825367 |
| Epigenetic therapies in combination with targeted therapies | ||||
| Azacitidine + milademetan | DNMT + MDM2 | 1 | 2014 | NCT02319369 |
| Decitabine + rapamycin or ribavirin | DNMT + mTOR | 1/2 | 2014 | NCT02109744 |
| Decitabine + BI836858 | DNMT + CD33 | 2 | 2015 | NCT02632721 |
| Decitabine + BP1001 | DNMT + Grb-2 | 2 | 2016 | NCT02781883 |
| Azacitidine + gilteritinib (ASP2215) | DNMT + FLT3 | 2/3 | 2016 | NCT02752035 |
| Decitabine + talazoparib | DNMT + PARP | 1/2 | 2016 | NCT02878785 |
| Decitabine + onvansertib | DNMT + PLK1 | 1b/2 | 2017 | NCT03303339 |
| Azacitidine + AZD2811 nanoparticles | DNMT + AURKB | 2 | 2017 | NCT03217838 |
| Azacitidine + SL-401 | DNMT + IL3 | 1 | 2017 | NCT03113643 |
| Decitabine + AMG-232 | DNMT + MDM2 | 1 | 2017 | NCT03041688 |
| Decitabine + pevonedistat (MLN4924) | DNMT + NEDD8 | 1 | 2017 | NCT03009240 |
| Azacitidine + pevonedistat | DNMT + NEDD8 | 3 | 2017 | NCT03268954 |
| Decitabine + venetoclax (ABT-199) | DNMT + BCL-2 | 2 | 2018 | NCT03404193 |
| Azacitidine + pevonedistat | DNMT + NEDD8 | 2 | 2018 | NCT03709576 |
| Azacitidine + HMPL-523 | DNMT + SYK | 1 | 2018 | NCT03483948 |
| Decitabine + quizartanib (AC-220) | DNMT + FLT3 | 1/2 | 2018 | NCT03661307 |
| Azacitidine + enasidenib mesylate (AG-221 mesylate) | DNMT + IDH2 | 2 | 2018 | NCT03683433 |
| Azacitidine + nintedanib (BIBF-1120) | DNMT + VEGF + FGFR + PDGFR | 1 | 2018 | NCT03513484 |
| Belinostat (PCD-101) + pevonedistat (MLN4924) | HDAC + NEDD8 | 1 | 2018 | NCT03772925 |
| Azacitidine + glasdegib (PF-04449913) | DNMT + SHH | 3 | 2018 | NCT03416179 |
| Pracinostat + gemtuzumab ozogamicin | HDAC + CD33 | 1 | 2019 | NCT03848754 |
| Azacitidine or decitabine + venetoclax (ABT-199) | DNMT + BCL-2 | 3 | 2019 | NCT03941964 |
| Azacitidine + APR-246 | DNMT + P53 | 2 | 2019 | NCT03931291 |
| Drug | Targets | Phase | Date | Clinical trial |
|---|---|---|---|---|
| Combinations of epigenetic therapies | ||||
| Azacitidine + entinostat (MS275) | DNMT + HDAC | 2 | 2011 | NCT01305499 |
| Azacitidine + FT-2102 | DNMT1 + IDH1 | 1/2 | 2016 | NCT02719574 |
| Azacitidine + LSD1 inhibitor (NCB059872) | DNMT1 + LSD1 | 1/2 | 2016 | NCT02712905 |
| Azacitidine + ivosidenib (AG-120) | DNMT + IDH1 | 3 | 2017 | NCT03173248 |
| Azacitidine + pracinostat (SB939) | DNMT + HDAC | 3 | 2017 | NCT03151408 |
| Decitabine + vorinostat before or during FLAG | DNMT + HDAC | 1 | 2017 | NCT03263936 |
| Azacitidine + PRMT5 inhibitor (GSK3326595) | DNMT + PRMT5 | 1 | 2018 | NCT03614728 |
| Low-dose azacitidine + vorinostat after alloHSCT | DNMT + HDAC | 1 | 2019 | NCT03843528 |
| Epigenetic therapies in combination with immunotherapies | ||||
| Azacitidine + nivolumab (MDX-1106) +/or ipilimumab (MDX-010) | DNMT + PD-1 + CTLA-4 | 2 | 2015 | NCT02397720 |
| Azacitidine + pembrolizumab (MK-3475) | DNMT + PD-1 | 2 | 2016 | NCT02845297 |
| Decitabine + ipilimumab (MDX-010) | DNMT + CTLA-4 | 1 | 2016 | NCT02890329 |
| Guadecitabine + atezolizumab (MPDL 3280A) | DNMT + PD-L1 | 1/2 | 2016 | NCT02935361 |
| Decitabine + PDR001 +/or MBG453 | DNMT + PD-1 + TIM-3 | 1 | 2017 | NCT03066648 |
| Azacitidine + Hu5F9-G4 | DNMT + CD47 | 1 | 2017 | NCT03248479 |
| Decitabine + CDX-1401 + poly ICLC + nivolumab (MDX-1106) | DNMT + DEC-205 + TLR-3 + PD-1 | 1 | 2017 | NCT03358719 |
| Decitabine + avelumab | DNMT + PD-L1 | 1 | 2018 | NCT03395873 |
| Azacitidine + nivolumab (ADVL1412) | DNMT + PD-1 | 1/2 | 2019 | NCT03825367 |
| Epigenetic therapies in combination with targeted therapies | ||||
| Azacitidine + milademetan | DNMT + MDM2 | 1 | 2014 | NCT02319369 |
| Decitabine + rapamycin or ribavirin | DNMT + mTOR | 1/2 | 2014 | NCT02109744 |
| Decitabine + BI836858 | DNMT + CD33 | 2 | 2015 | NCT02632721 |
| Decitabine + BP1001 | DNMT + Grb-2 | 2 | 2016 | NCT02781883 |
| Azacitidine + gilteritinib (ASP2215) | DNMT + FLT3 | 2/3 | 2016 | NCT02752035 |
| Decitabine + talazoparib | DNMT + PARP | 1/2 | 2016 | NCT02878785 |
| Decitabine + onvansertib | DNMT + PLK1 | 1b/2 | 2017 | NCT03303339 |
| Azacitidine + AZD2811 nanoparticles | DNMT + AURKB | 2 | 2017 | NCT03217838 |
| Azacitidine + SL-401 | DNMT + IL3 | 1 | 2017 | NCT03113643 |
| Decitabine + AMG-232 | DNMT + MDM2 | 1 | 2017 | NCT03041688 |
| Decitabine + pevonedistat (MLN4924) | DNMT + NEDD8 | 1 | 2017 | NCT03009240 |
| Azacitidine + pevonedistat | DNMT + NEDD8 | 3 | 2017 | NCT03268954 |
| Decitabine + venetoclax (ABT-199) | DNMT + BCL-2 | 2 | 2018 | NCT03404193 |
| Azacitidine + pevonedistat | DNMT + NEDD8 | 2 | 2018 | NCT03709576 |
| Azacitidine + HMPL-523 | DNMT + SYK | 1 | 2018 | NCT03483948 |
| Decitabine + quizartanib (AC-220) | DNMT + FLT3 | 1/2 | 2018 | NCT03661307 |
| Azacitidine + enasidenib mesylate (AG-221 mesylate) | DNMT + IDH2 | 2 | 2018 | NCT03683433 |
| Azacitidine + nintedanib (BIBF-1120) | DNMT + VEGF + FGFR + PDGFR | 1 | 2018 | NCT03513484 |
| Belinostat (PCD-101) + pevonedistat (MLN4924) | HDAC + NEDD8 | 1 | 2018 | NCT03772925 |
| Azacitidine + glasdegib (PF-04449913) | DNMT + SHH | 3 | 2018 | NCT03416179 |
| Pracinostat + gemtuzumab ozogamicin | HDAC + CD33 | 1 | 2019 | NCT03848754 |
| Azacitidine or decitabine + venetoclax (ABT-199) | DNMT + BCL-2 | 3 | 2019 | NCT03941964 |
| Azacitidine + APR-246 | DNMT + P53 | 2 | 2019 | NCT03931291 |
A list of currently active clinical trials that involve epigenetic therapies in combination with targeted therapies, immunotherapies, and/or other epigenetic therapies for the treatment of AML.
alloHSCT, allogeneic hematopoietic stem cell transplantation; AURKB, Aurora kinase B; BCL-2, B-cell lymphoma 2; DNMT, DNA methyltransferase; FGFR, fibroblast growth factor receptor; Grb2, growth factor receptor bound protein 2; HDAC, histone deacetylase; IDH, isocitrate dehydrogenase; IL3, interleukin 3; LSD1, lysine-specific histone demethylase 1; MDM2, mouse double minute 2; mTOR, mammalian target of rapamycin; PARP, poly ADP ribose polymerase; PD-1, programmed cell death protein 1; PDGFR, platelet-derived growth factor receptor; PD-L1, programmed cell death ligand 1; PLK1, polo-like kinase 1; PRMT5, protein arginine N-methyltransferase 5; Shh, Sonic hedgehog; TIM-3, T-cell immunoglobulin and mucin-domain containing-3; TLR-3, Toll-like receptor 3; VEGF, vascular endothelial growth factor.
IDH1 and 2 inhibitors
The cytosolic protein IDH1 and mitochondrial protein IDH2 function to produce a key intermediary metabolite of the citric acid cycle, α-ketoglutarate (α-KG), which is essential for cellular metabolism.29 Comprehensive sequencing studies have shown that ∼15% to 20% of AML genomes harbor mutations in IDH1 or 2 and these mutations are more prevalent in patients with a normal karyotype.2,3 Mutations occur largely within the active site and confer neomorphic activity, whereby the mutant enzymes convert α-KG into the oncometabolite, 2-hydroxyglutarate (2-HG).30 The levels of 2-HG can reach millimolar concentrations in mutant cells leading to competitive inhibition of many dioxygenases that are dependent on α-KG as a substrate.31,32 In particular, 2-HG–mediated inhibition of the TET family of DNA demethylases and the Jumonji family of histone demethylases leads to widespread DNA and histone hypermethylation, respectively, which is associated with altered gene expression.10,32,33 The effects of IDH1/2 mutations largely phenocopy TET2 loss-of-function mutations and these mutations are invariably mutually exclusive in patients, raising the possibility that IDH1/2 mutations drive malignancy primarily by disrupting the function of TET2.10 It must be noted, however, that although IDH1/2 and TET2 share a common pathway, AML with IDH1/2 mutations are functionally and clinically distinct from TET2-mutated AML, suggesting that they exhibit TET-independent activity that also contributes to their role in leukemogenesis.
How the specific changes in DNA or histone methylation instigated by IDH1/2 mutations contribute to the pathogenesis of AML remains largely unknown. Hypermethylation is observed not only at CpG islands of promoters, where it can directly silence tumor-suppressor genes, but also at other regulatory elements, such as enhancers and insulators. Many TFs are sensitive to DNA methylation and therefore hypermethylation could disrupt binding of TFs that recognize CG-rich motifs.34 One such TF is CTCF, which is an architectural protein critically important for the establishment and maintenance of topologically associated domains (TADs). TADs are a fundamental organization unit of the genome and are thought to prevent promiscuous, ectopic interaction of enhancers with unrelated promoters.35,36 Interestingly in glioma, IDH mutations have been shown to trigger hypermethylation of CTCF-binding sites leading to oncogene activation, as promoters and enhancers that are normally insulated from each other due to localization in different TADs undergo promiscuous interactions.37 It is reasonable to suspect that IDH mutations in AML could lead to similar molecular consequences. Further characterization of chromatin topology in this context may help explain oncogenic gene-expression programs that currently cannot be directly correlated with methylation changes in gene promoters. Additionally, it is likely that the widespread hypermethylation observed at enhancer elements could disrupt the function of other TFs, which may contribute to the transcriptional dysregulation observed in AML.
The neomorphic function of the mutant IDH enzymes provides an attractive target for small molecule intervention and has motivated the pharmaceutical industry to develop specific inhibitors for the mutant form of both enzymes. Small molecule inhibition of IDH1/2 results in reduction of 2-HG levels, reversal of DNA and histone hypermethylation, and a release on the differentiation block caused by these mutations.38-40 Durable remissions with enasidenib (IDH2 inhibitor) and ivosidenib (IDH1 inhibitor) can be achieved in ∼20% to 30% of patients with relapsed/refractory IDH1/2 mutant AML and, as a consequence, these therapies have been approved by the FDA.39,41-43 Although these drugs may offer patients with IDH-mutant AML a significant improvement in clinical outcome, resistance is nonetheless invariably seen. Increased mutational burden or the presence of co-occurring NRAS mutations appear to be indicators of primary resistance to enasidenib therapy, suggesting that other non-IDH containing clones need to be eliminated to achieve better responses.44 Additionally, potent small molecule inhibition places enormous evolutionary pressure on the malignant cell population and can favor the outgrowth of a clone harboring a resistance conferring mutation. This has already been identified with IDH inhibitors whereby acquired resistance can arise due to isoform switching from mutant IDH1 to mutant IDH2 and vice versa or through clonal evolution in the absence of second-site IDH mutations.45-47
Epigenetic therapies currently in clinical trials
Epigenetic compounds targeting transcriptional dependencies in AML
The realization that epigenetic dysregulation underpins the pathogenesis of AML has led to a number of unbiased genetic and chemical screens aimed at identifying critical dependencies and consequently novel therapeutic targets for this disease. These efforts have led to the identification of key targets and the subsequent development of a number of potent small molecules that inhibit the function of the epigenetic writers (disruptor of telomeric silencing 1-like [DOT1L], protein arginine N-methyltransferase 5 [PRMT5]), epigenetic erasers (lysine-specific histone demethylase 1 [LSD1]), and epigenetic readers (the bromodomain and extra-terminal motif [BET] bromodomain proteins, including BRD4).48-57 Importantly, all of these drug targets are not frequently mutated or overexpressed in AML, instead they function as key nononcogene dependencies to sustain the malignant transcription program of leukemic cells.52-55 The preclinical evaluation of these newer compounds showed striking leukemia-specific efficacy in a range of AML models in vitro and in vivo. Based on the success of these small molecules in the laboratory, drugs targeting DOT1L, PRMT5, LSD1, and BRD4 have transitioned into early-phase clinical trials in AML (Figure 1). Although some patients have achieved durable remissions, the emerging data from these clinical trials have not paralleled the preclinical excitement generated by these drugs.6,58
Why have the preclinical results with epigenetic therapies not approximated their clinical outcomes?
It is interesting to speculate about the potential reason for the discrepancy between the dramatic results of epigenetic therapies in preclinical studies and the more modest clinical outcomes achieved thus far. First, it is important to appreciate that as a consequence of clinical trial design, all of these epigenetic therapies have been trialed primarily in patients with relapsed, refractory cancers that have been exposed to multiple rounds of therapeutic selection; few, if any, preclinical assays model this complex clinical scenario.6 Another major difference between preclinical models and patients in the clinic is that preclinical studies that are performed in either syngeneic or patient-derived xenograft models rarely replicate the genetic and transcriptional heterogeneity seen in human AML. Moreover, in contrast to the clinical environment, these preclinical studies are conducted in a germ-free environment and, in the case of patient-derived xenograft models, in immunodeficient hosts. The importance of these genetic and environmental factors and their influence on epigenetic regulation should not be underestimated; the epigenome in tumor cells and cells within the tumor microenvironment is highly dynamic and is constantly adapting to metabolic, therapeutic, and other environmental pressures.59 Although the initial clinical trials with these epigenetic therapies may be perceived by some as being disappointing, recall that no single-agent therapy, including those that target signaling pathways, apoptotic machinery, or antibodies against cell surface proteins, has produced significant cures in AML; the cornerstone of curative therapy in AML is combination therapy.
Combination therapies with epigenetic agents
The success of epigenetic therapies for the treatment of AML (and indeed other cancers) depends on the identification and implementation of rational drug combinations. An important question that requires urgent attention and clarification is how best to combine epigenetic drugs and with what other classes of drugs? The options for combination strategies include pairing epigenetic therapies with chemotherapy, targeted therapy (including other epigenetic therapies), and/or with immunotherapy. Identifying the most effective combination strategies will take time and careful evaluation, especially given that the specific genetic and/or epigenetic state of the tumor will undoubtedly influence the outcome. The major currently active clinical trials assessing combination strategies incorporating epigenetic therapies in AML are summarized in Table 1.
Combinations of epigenetic therapies
Initial clinical studies in AML/myelodysplastic syndrome combining 2 histone deacetylase (HDAC) inhibitors and DNA-hypomethylating agents suggested some promise.60 However, randomized studies with these agents failed to show any benefit61 and, more concerning in some cases, they appeared to be functionally antagonistic.62 This functional antagonism serves to highlight the potential dangers of an empirical clinical approach to combination strategies involving epigenetic agents. Given the complexity of epigenetic regulation, it is difficult to predict which combinations of epigenetic agents are likely to demonstrate synergy. Many, if not most, epigenetic proteins contribute to several different chromatin complexes with each protein complex performing a context-dependent function. Therefore, an important limitation of current epigenetic therapies is the broad inhibition of all complexes containing the target protein, potentially generating unwanted adverse effects and in some cases unexpected outcomes. This reinforces the importance of a thorough molecular understanding prior to clinical combination, as has been performed for AML with mixed lineage leukemia (MLL) fusions63,64 and TET2 mutations.65 Some of these combination strategies including those that combine LSD1 or PRMT5 inhibitors with DNA-hypomethylating agents have now started to progress to clinical trials (Table 1).
Combination of epigenetic drugs with immunotherapies
There is now a large body of accumulating evidence highlighting the importance of the functional interplay between epigenetic therapies and an intact immune system.66,67 The importance of an intact immune system for the efficacy of many epigenetic drugs is highlighted by the different treatment outcomes observed when identical tumors are treated with epigenetic therapies in an immune-competent vs immune-deficient hosts.68,69 In addition to hypomethylating agents, a number of other epigenetic drugs, such as inhibitors of EZH2 and LSD1 can induce ERV reexpression, resulting in a state of “viral mimicry” that contributes to their efficacy26,70,71 (Figure 3). Similarly, BET inhibitors have been shown to inhibit the expression of immune-checkpoint molecules such as PD-L1, which enhances anticancer immune surveillance69 (Figure 3). In fact, a common theme emerging from clinical trials with epigenetic therapies (and the longstanding clinical experience with DNA-hypomethylating agents) is the fact that these therapies do not have an immediate cytotoxic effect. Consistent with a role for requiring the immune system, most patients treated with epigenetic therapies have their maximum therapeutic benefit several months after commencing the drug.39,48,72,73 These clinical observations and the accumulating preclinical evidence supporting the importance of the immune system in facilitating the efficacy of epigenetic therapies has fueled the clinical evaluation of various epigenetic therapies in combination with cancer immunotherapies. The importance of the functional interaction between epigenetic therapies and anticancer immune surveillance has recently been reviewed74 and we have summarized the major combination therapy studies in AML in Table 1.
Future epigenetic therapies in AML: disrupting a leukemia-specific transcriptional program
Although further characterization and optimization of the current generation of epigenetic drugs is certainty necessary, 1 notable limitation of the current generation of drugs is that they all target ubiquitously expressed chromatin regulators often leading to unintended consequences. Therefore, more dramatic improvements may come from new therapies that directly target lineage-specific oncogenic dependencies that are used by the majority of AML subsets to maintain the malignant transcription program.
TFs as viable therapeutic targets
How may one approach the therapeutic targeting of the majority of cases of AML? In this regard, TFs represent an ideal target for specifically disrupting oncogenic transcriptional programs in specific cell lineages. TFs sit at the heart of the cellular regulatory networks and drive malignant cell transcriptional programs by acting as guideposts for epigenetic complexes, recruiting them to regulatory elements in a sequence-specific manner.75,76 In mammals, TFs comprise ∼10% of the protein-coding genome and outnumber the epigenetic complexes they recruit.77 As a result, numerous TFs must converge on a limited number of epigenetic complexes, which then act as tools to exert locus-specific functions. Therefore, despite some clear examples of tissue-specific functions, in general, epigenetic complexes display less specificity than lineage-restricted TFs.78,79 Because specificity directly relates to therapeutic window, this highlights the clear theoretical benefit of targeting TFs over epigenetic complexes. In support of this idea, inspection of the dependencies across various AML subtypes highlights MYB, PU.1, CEBPA, and RUNX1 as some of the strongest and most specific AML dependencies.80,81
Although targeting TFs represents an exciting strategy for disrupting specific oncogenic transcriptional programs, it is inherently difficult to design molecules to directly disrupt their function. Due to their protein structure, most TFs do not have an easily identifiable druggable domain. Recently, a number of alternative approaches have emerged that attempt to circumvent these current limitations, in the hope of disrupting a more leukemia-specific program.
Blocking interactions between TFs and epigenetic complexes
Although the general transcription machinery is broadly considered to be ubiquitously expressed and generic in its function, a number of recent publications suggest that, in some instances, there is a locus-specific requirement for particular subunits due to specific interactions with TFs.77,82,83 Therefore, targeting these subunit-TF interactions may provide an opportunity to disrupt more specific oncogenic transcriptional programs. A proof of principle was provided recently for the oncogenic TF MYB, which is a common dependency in a range of AML cell lines. Much like other TFs, MYB is difficult to target directly due to the lack of a clear functional pocket amenable to small molecule inhibition. To circumvent this, the Vakoc laboratory disrupted a TATA-box binding protein associated factor 12 (TAF12)-dependent interaction between MYB and the TFIID complex using a “squelching peptide,” which resulted in a marked leukemia-specific antiproliferative effect without any other overt phenotypic consequences.84 In addition to its interaction with TAF12, MYB has been shown to interact with the KIX domain of CREB-binding protein (CBP)/P300, an interaction that is amenable to pharmacological inhibition (Figure 4A).85-87 Therefore, interrupting interactions between oncogenic TFs and particular subunits of large epigenetic complexes has the potential to specifically disrupt oncogenic transcriptional programs, circumventing the issues associated with targeting TFs directly. However, given that many of these interactions between TFs and epigenetic complexes are driven by “fuzzy” interactions between low-complexity domains, the degree of specificity between TFs and a particular epigenetic domains/subunit remains unclear. The absence of a structured lock-and-key mechanism has restricted our ability to understand the rules of engagement between low-complexity domains, ultimately limiting our capacity to therapeutically exploit these types of interactions. In fact, it is yet to be determined whether small molecules can even be developed to target these interactions. These outstanding questions must be addressed if we hope to proceed with this potentially promising therapeutic strategy.
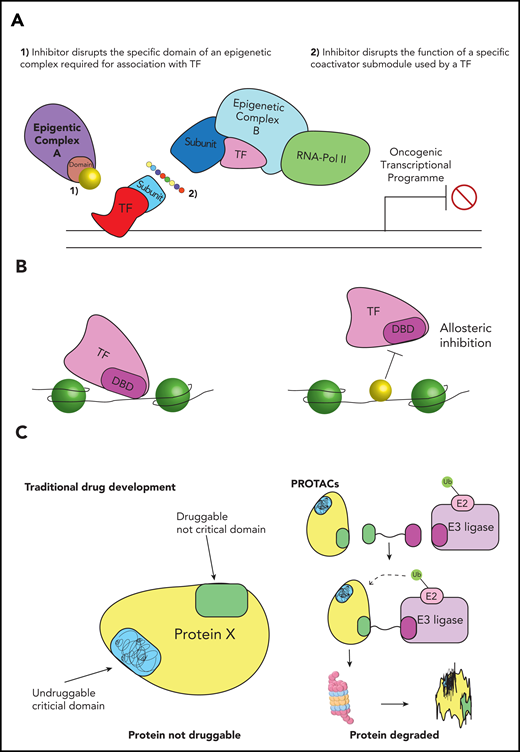
Developing more specific targeted therapies for AML. (A) Disrupting specific epigenetic programs. Targeting TFs would enable disruption of more specific oncogenic programs, however, drugs against TFs are difficult to develop. To circumvent this limitation, new therapies could either (1) block specific domains on generic epigenetic proteins that interact with an oncogenic TF or (2) disrupt specific submodules of large epigenetic complexes, which are recruited by a particular TF. Either of these approaches could retain druggability, while also disrupting a more AML-specific program. (B) Disrupting DNA binding of epigenetic proteins. TFs and epigenetic proteins can be recruited to DNA by specific DNA sequences. Inhibitors that interact with these DNA sequences could potentially block binding of a TF or epigenetic protein, resulting in disruption of a more AML-specific transcriptional program. (C) Degradation of epigenetic proteins or oncogenic drivers. Traditional drug development requires the functional domain of a target protein to have a structure amenable to inhibition. Proteolysis-targeting chimera (PROTAC)-based degradation strategies do not require the specific functional domain to be druggable; instead small molecules or peptides can be designed against any region of the protein of interest. These molecules are fused to a linker that brings the protein into close proximity with an E3 ubiquitin ligase. This proximity causes ubiquitination, resulting in its degradation of the target protein. This is likely to dramatically expand the number of oncogenic proteins that are druggable. DBD, DNA-binding domain.
Developing more specific targeted therapies for AML. (A) Disrupting specific epigenetic programs. Targeting TFs would enable disruption of more specific oncogenic programs, however, drugs against TFs are difficult to develop. To circumvent this limitation, new therapies could either (1) block specific domains on generic epigenetic proteins that interact with an oncogenic TF or (2) disrupt specific submodules of large epigenetic complexes, which are recruited by a particular TF. Either of these approaches could retain druggability, while also disrupting a more AML-specific program. (B) Disrupting DNA binding of epigenetic proteins. TFs and epigenetic proteins can be recruited to DNA by specific DNA sequences. Inhibitors that interact with these DNA sequences could potentially block binding of a TF or epigenetic protein, resulting in disruption of a more AML-specific transcriptional program. (C) Degradation of epigenetic proteins or oncogenic drivers. Traditional drug development requires the functional domain of a target protein to have a structure amenable to inhibition. Proteolysis-targeting chimera (PROTAC)-based degradation strategies do not require the specific functional domain to be druggable; instead small molecules or peptides can be designed against any region of the protein of interest. These molecules are fused to a linker that brings the protein into close proximity with an E3 ubiquitin ligase. This proximity causes ubiquitination, resulting in its degradation of the target protein. This is likely to dramatically expand the number of oncogenic proteins that are druggable. DBD, DNA-binding domain.
Disrupting recruitment at the DNA interface
For the most part, epigenetic proteins are targeted to chromatin by TFs, through highly conserved DNA-binding domains. Blocking the interaction between the TF and DNA may therefore be a viable strategy to disrupt specific oncogenic transcriptional programs. A new approach to blocking these interactions was recently shown for the critical AML TF, Pu.1. The Steidl group sought to disrupt the function of Pu.1 by blocking its interaction at the DNA interface.88 By using a small molecule inhibitor that specifically interacts with DNA at the AT-rich tracks within the minor groove at Pu.1 motifs, they were able to block binding to the major groove via allosteric inhibition (Figure 4B). Importantly, the inhibition of Pu.1 binding abrogated cell growth of AML cell lines with minimal toxicity to bone marrow cells. Although other examples of drugs that disrupt TF binding have been reported, the specificity and broad applicability of this approach requires further interrogation.89
Expanding the therapeutic landscape with targeted degradation strategies
Traditional drug development utilizes small molecules to block specific functional domains. The identity and/or structure of these functional domains in some potential targets are often unsolved or in other instances, the functional regions are unstructured, limiting the scope for drug development. In particular, TFs and coactivator proteins have highly unstructured domains that appear to be key to their function.90,91 The prospect of targeting these “undruggable” proteins has been approached by a number of groups that have developed tools that co-opt the endogenous ubiquitin ligase machinery to degrade proteins of interest. These proteolysis-targeting chimera (PROTAC) molecules use heterobifunctional ligands, to bring the target protein into close proximity to ubiquitin ligases, such as cereblon or VHL, targeting them for proteosomal degradation.92-95 Because these methods only require tethering of the target protein to the ubiquitin ligase and do not require inhibition of a functional domain, small molecules can be designed against any region of the protein of interest (Figure 4C). With future refinement, this approach has the potential to dramatically expand the number of TFs and epigenetic proteins that can be disrupted, allowing more specific leukemia vulnerabilities to become viable therapeutic targets. The significant promise of PROTACs is currently limited by their poor stability, bioavailability, and tissue/cell penetration.96 Once these obstacles are overcome, it will be vital to ensure specificity of degron recruitment. As the major target substrate is degraded and the effective concentration of the free small molecule increases, there is the possibility of lower affinity off-target interactions with other proteins becoming substrates for degradation.97 This in turn may potentially lead to profound, undesired off-target effects.
Conclusions
Aberrant chromatin landscapes and nononcogenic epigenetic dependencies play a central role in the initiation and maintenance of AML. Overcoming these dysregulated gene-expression programs through epigenetic therapies remains an important therapeutic strategy. The current modest clinical effects seen with this class of drugs may in part reflect imperfect preclinical modeling and a limited molecular understanding of the strengths and weaknesses of the current generation of compounds.98 Nevertheless, it should be remembered that epigenetic regulators function primarily to nuance access to the DNA template for DNA repair, replication, and gene expression. They are not classical cytotoxic agents and tumor lysis syndrome has not been reported as yet for these agents, even in patients with a large burden of disease. Many of these drugs take months to exert their maximum effect; emerging evidence suggests that the immune system may play a critical role in their clinical activity. Lessons learned from the current generation of epigenetic therapies will unquestionably pave the way for appropriate combination strategies and pioneering new methods to more specifically disrupt oncogenic dependencies that drive AML. With better preclinical modeling, well-informed drug combinations, and enhanced precision for targeting epigenetic regulators, we envisage a bright future in which epigenetic drugs will ultimately be used in combination with other therapies as an effective strategy to overcome transcriptional dysregulation in AML.
Authorship
Contribution: K.A.F., C.C.B., and M.A.D. wrote the manuscript and approved the final version.
Conflict-of-interest disclosure: M.A.D. has been a member of advisory boards for CTX CRC, Storm Therapeutics, Celgene, Cambridge Epigenetix, and GlaxoSmithKline. The Dawson laboratory receives research funding from Pfizer. The remaining authors declare no competing financial interests.
Correspondence: Mark A. Dawson, Cancer Epigenetics Laboratory and Department of Haematology, Peter MacCallum Cancer Centre, 305 Grattan St, Melbourne, VIC 3000, Australia; e-mail: mark.dawson@petermac.org.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal