

Key Points
During bradykinin-driven inflammation, C1-inhibitor is too weak to control the contact system enzymes.
Redesigned α1-antitrypsin variants attenuate bradykinin-driven inflammatory reactions and thrombosis in vivo.

Abstract
The contact system produces the inflammatory peptide bradykinin and contributes to experimental thrombosis. C1 esterase-inhibitor (C1INH) deficiency or gain-of-function mutations in factor XII (FXII) cause hereditary angioedema, a life-threatening tissue swelling disease. C1INH is a relatively weak contact system enzyme inhibitor. Although α1-antitrypsin (α1AT) does not naturally inhibit contact system enzymes, a human mutation (M358R; α1AT-Pittsburgh) changes it into a powerful broad-spectrum enzyme inhibitor. It blocks the contact system, but also thrombin and activated protein C (APC), making it an unattractive candidate for therapeutic contact system blockade. We adapted the reactive center loop of α1AT-Pittsburgh (AIPR/S) to overcome these obstacles. Two α1AT variants (SMTR/S and SLLR/S) strongly inhibit plasma kallikrein, activated FXII, and plasmin. α1AT-SMTR/S no longer inhibits thrombin, but residually inhibits APC. In contrast, α1AT-SLLR/S residually inhibits thrombin, but no longer APC. Additional modification at the P1′ position (S→V) eliminates residual inhibition of thrombin and APC for both variants, while retaining their properties as contact system inhibitors. Both α1AT-SMTR/V and -SLLR/V are superior to C1INH in reducing bradykinin production in plasma. Owing to their capacity to selectively block contact system-driven coagulation, both variants block vascular occlusion in an in vivo model for arterial thrombosis. Furthermore, both variants block acute carrageenan-induced tissue edema in mice. Finally, α1AT-SLLR/V, our most powerful candidate, suppresses epithelial leakage of the gut in a mouse model of colitis. Our findings confirm that redesign of α1AT strongly alters its inhibitory behavior and can be used for the treatment of contact system-mediated thrombosis and inflammation.
Introduction
Serine protease inhibitors (SERPINs) are critical regulators of serine proteases.1 These SERPINs regulate important physiological processes such as coagulation, fibrinolysis, complement activation, inflammation, and angiogenesis.
C1 esterase-inhibitor (C1INH) is the main SERPIN for C1 esterase of the complement system. It also inhibits activated factor XII (FXIIa), plasma kallikrein (PKa), and factor XIa. These enzymes belong to the plasma contact system and the coagulation system.2 Individuals with C1INH deficiency develop attacks of tissue swelling in a disease called hereditary angioedema (HAE; C1INH-HAE). This is attributable to escalated release of the inflammatory mediator bradykinin from its precursor high-molecular-weight kininogen (HK) by PKa. There are no clinical signs of thrombosis in C1INH-deficient patients.3
C1INH is a relatively inefficient enzyme inhibitor compared with other SERPINs (supplemental Table 1, available on the Blood Web site).4-6 It is probable that this is required for a balanced physiological bradykinin production. Interestingly, bradykinin-driven HAE can also occur when plasma levels and C1INH activity are normal. Specific subtypes of HAE independent of C1INH deficiency are now recognized. These are mostly associated with gain-of-function mutations in factors that contribute to bradykinin production, such as FXIIa (FXII-HAE)7-9 or plasmin (PLG-HAE).10 High-dose C1INH administration is therapeutic for some patients with these forms of HAE.11,12 In many other cases, the underlying cause of HAE with normal C1INH is still unknown (U-HAE12 ). However, there is good evidence that bradykinin is the key inflammatory mediator in this setting.13
The role of bradykinin in inflammatory reactions is not limited to HAE. Bradykinin is also implicated in Alzheimer’s disease, colitis, arthritis, and sepsis despite normal C1INH activity.14-17 As a result, therapeutic strategies that target the molecular axis that produces bradykinin are of great interest.
SERPINs contain a conformationally strained reactive center loop (RCL), which is cleaved by target enzymes between the P1 and P1′ residues. This allows the cleaved RCL to rapidly insert into the SERPIN molecule that undergoes a conformational change. As a result, the active site of the bound protease becomes distorted before it can dissociate, trapping it in a covalent complex with the SERPIN.18 The amino acid sequence of the RCL is critical to SERPIN specificity, and changes in it can have dramatic effects. For example: a P1 point mutation (M358R) in human α1-antitrypsin (α1AT) changes its RCL sequence from AIPM/S to AIPR/S (spanning from P4 to P1′; "/" indicates the reactive center). Whereas wild-type α1AT (α1AT-WT) normally inhibits neutrophil elastase, α1AT-Pittsburgh constitutes a broad-spectrum inhibitor of coagulation enzymes, including thrombin.6 This alteration results in bleeding episodes in patients with α1AT-Pittsburgh.19 After its discovery, recombinant α1AT-Pittsburgh was investigated in a baboon model sepsis-associated coagulopathy, but without success.20 Here, α1AT-Pittsburgh infusion caused increased lethality, which remains largely unexplained. In unrelated studies, it was found that α1AT-Pittsburgh has the ability to block activated protein C (APC).21 APC controls coagulation FVa and FVIIIa and protects against venous thrombosis.22,23 Building on this observation, a refined, more selective, APC-blocking α1AT variant (KR/K) is currently under development as a therapy for hemophilia.24
α1AT-Pittsburgh is a very competent inhibitor of FXIIa, PKa, and plasmin (8.3-, 5.2-, and 250-fold more powerful than C1INH, respectively).6 Moreover, α1AT is less glycosylated than C1INH and has a longer half-life (208 vs 32 hours).25,26 These properties make it an attractive candidate for therapeutic inhibition of contact system enzymes. However, its ability to block thrombin and FXa critically limits its suitability as a therapeutic agent, as treatment with this molecule is likely to cause bleeding adverse effects. The original patient with α1AT-Pittsburgh had a lifelong bleeding state and died at the age of 7 years because of hemorrhage.19 We here report on the development and characterization of 2 novel α1AT-Pittsburgh variants with redesigned RCLs for treatment of contact system-driven thrombosis and inflammation.
Methods
Detailed methods for protein production and characterization are provided in the supplemental Data.27–31
Performance of α1-antitrypsin variants in in vivo models for thrombosis and inflammation
Carrageenan-induced paw swelling.
Paw swelling studies32,33 were performed by ImmunoPrecise Antibodies (Victoria, Canada) in a blinded manner. Ethical approval was obtained from the Canadian Council on Animal Care, and all work was performed according to their standards. Female BALB/C mice (8 weeks old, 12 mice per group) received 100-μL IV tail vein injections with 8 mg/kg α1AT variant, 16 mg/kg rC1INH, 1 mg/kg HOE140 (icatibant), or 5 mg/kg indomethacin in sterile PBS. Thirty minutes later, carrageenan (20 μL, 1% wt/vol in saline) was injected in the dorsal part of the left hind paw of each mouse. Changes in paw size were measured in triplicate by a digital micrometer34 at 0 and 10 minutes and 2, 4, and 6 hours after carrageenan injection.
Dextran sulfate-induced colitis.
Experimental colitis studies were performed in a blinded manner by Preclinics (Potsdam, Germany) under local ethical approval (2347-29-2017 Ä1, Land Brandenburg LAVG). Female NMRI mice (7 weeks old, 6 mice per group) were acclimatized for 2 weeks on a Ssniff R/M-H Etrudat diet and drinking water ad libitum. From day 0 onward, the drinking water was replaced with water containing 3% (wt/vol) dextran sulfate sodium. On day 0 and 3, mice received 200 μL IV injections (tail vein) with either 8 mg/kg α1AT variant or 16 mg/kg rC1INH in sterile PBS. On day 6, mice received a 5 mL/kg bodyweight gavage of a fluorescein isothiocyanate (FITC)–dextran solution (4 kDa, 120 mg/mL). Four hours later, they were anesthetized via 5% isoflurane in O2 and exsanguinated. Blood was collected in heparin-lithium for plasma isolation. The FITC signal in the plasma was quantified in a 96-well plate by fluorescence measurements (Ext:485nm, Em:535nm). A FITC-dextran standard curve was included for quantification.
FeCl3-induced arterial thrombosis.
Arterial thrombosis studies were performed at the animal facilities of Hamburg-Eppendorf (Germany) under local ethical approval (Behörde für Gesundheit und Verbraucherschutz Freie und Hansestadt Hamburg; #N56/2018) in a blinded manner.35 C57BL/6 mice (8 weeks old, 7 mice per group) were anesthetized by inhalation of 4% isoflurane and continuously maintained at 1.5% isoflurane. A flow probe (Transonic systems; TS420) was fitted around the exposed artery to monitor blood flow. Mice received 200 μL IV tail vein injections (8 mg/kg α1AT variant, 16 mg/kg rC1INH in sterile PBS) and 100 μL subcutaneous Metamizol (200 mg/kg) for pain relief. After 30 minutes, FeCl3 (5% wt/vol) was applied topically to the exposed artery via a piece of filter paper (1 × 1.5 mm) to induce thrombus formation. After 3 minutes, the filter paper was removed while blood flow was continuously recorded until cessation of blood flow or, alternatively, up to 40 minutes.
Results
Modification of α1-antitrypsin Pittsburgh’s reactive center loop for contact system enzyme blockade
We set out to improve and refine the specificity of α1AT-Pittsburgh toward enzymes that are involved in bradykinin production (ie, FXIIa, PKa, and plasmin). As the P1 arginine (R358) is essential for this function,36 we replaced the N-terminally positioned P4-P2 amino acids with new sequences (Figure 1A). We first reasoned that the activation loop of FXII might be an attractive sequence donor: this sequence can be cleaved by FXIIa, PKa, and FXIa, as well as plasmin after R353, resulting in FXII activation.37,38 In contrast, thrombin, FXa, or APC cannot cleave FXII at this position (supplemental Figure 1A). Accordingly, we developed the α1AT variant SMTR/S (Figure 1A). In a complementary approach, we performed an in silico screen of previously published tri-peptide substrate libraries for potential sequence donors.39,40 We scored the reactivity of selected enzymes toward synthetic peptide substrates as poor (0 points), intermediate (1 point), or good (2 points) to develop a predictive model. Substrates were subsequently ranked by overall score. Scores for conversion by thrombin, APC, FXIa, FXa, FIXa, and FVIIa (undesirable) were subtracted from scores for conversion by FXIIa and PKa (desirable; supplemental Figure 1B-C). In this way, we developed 18 α1AT variants and functionally screened their inhibitory properties (at fixed SERPIN concentrations) in cell culture supernatant (supplemental Table 5). Subsequently, we selected α1AT variant SLLR/S as the most promising candidate (Figure 1A). We then produced and purified α1AT-SMTR/S and α1AT-SLLR/S, alongside α1AT-WT and α1AT-Pittsburgh (as controls), to investigate their inhibitory properties and specificity in comparison with plasma-derived C1INH (pdC1INH).
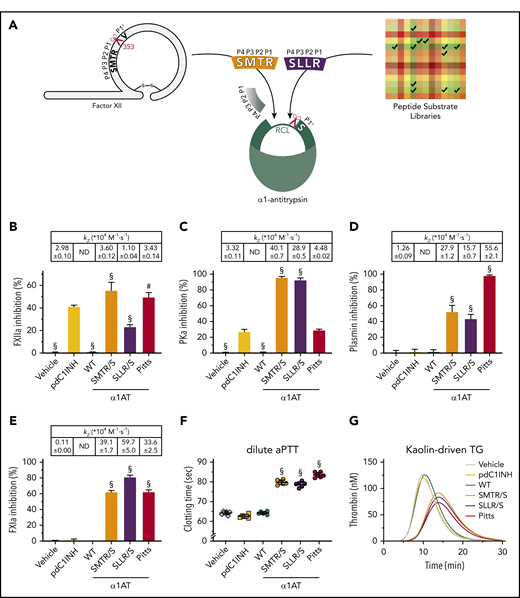
Modification of α1-antitrypsin Pittsburgh’s RCL for contact system enzyme blockade. (A) α1-Antitrypsin reactive center loop grafting strategy. The reactive center is indicated by a scissors symbol. (B-E) Screening of target enzymes inhibition by α1AT variants and their controls; table insets show second-order rate constants (k2: 104 M−1·s−1; ND = not detected). (B) Inhibition of 25 nM αFXIIa by 96.2 nM SERPIN. (C) Inhibition of 2.3 nM PKa by 28.9 nM SERPIN. (D) Inhibition of 24.1 nM plasmin by 24.1 nM SERPIN. (E) Inhibition of 2.5 nM FXIa by 9.6 nM SERPIN. (F) Inhibition of dilute aPTT clotting times in normal plasma by 384 nM SERPIN or buffer (vehicle). (G) Inhibition of kaolin-driven thrombin generation in normal plasma by 384 nM SERPIN or buffer (vehicle). Data represent the mean ± standard deviation (SD) of 3 separate experiments, each performed in duplicate. #P < .005; §P < .0001, compared with pdC1INH by 1-way analysis of variance (ANOVA). pdC1INH, plasma-derived C1INH; Pitts, α1AT-Pittsburgh; SLLR/S, α1AT-SLLR/S; SMTR/S, α1AT-SMTR/S; WT, wild-type α1AT.
Modification of α1-antitrypsin Pittsburgh’s RCL for contact system enzyme blockade. (A) α1-Antitrypsin reactive center loop grafting strategy. The reactive center is indicated by a scissors symbol. (B-E) Screening of target enzymes inhibition by α1AT variants and their controls; table insets show second-order rate constants (k2: 104 M−1·s−1; ND = not detected). (B) Inhibition of 25 nM αFXIIa by 96.2 nM SERPIN. (C) Inhibition of 2.3 nM PKa by 28.9 nM SERPIN. (D) Inhibition of 24.1 nM plasmin by 24.1 nM SERPIN. (E) Inhibition of 2.5 nM FXIa by 9.6 nM SERPIN. (F) Inhibition of dilute aPTT clotting times in normal plasma by 384 nM SERPIN or buffer (vehicle). (G) Inhibition of kaolin-driven thrombin generation in normal plasma by 384 nM SERPIN or buffer (vehicle). Data represent the mean ± standard deviation (SD) of 3 separate experiments, each performed in duplicate. #P < .005; §P < .0001, compared with pdC1INH by 1-way analysis of variance (ANOVA). pdC1INH, plasma-derived C1INH; Pitts, α1AT-Pittsburgh; SLLR/S, α1AT-SLLR/S; SMTR/S, α1AT-SMTR/S; WT, wild-type α1AT.
In comparison with pdC1INH, α1AT-SMTR/S and α1AT-Pittsburgh are slightly stronger inhibitors of FXIIa, whereas α1AT-SLLR/S is slightly weaker (Figure 1B; table inset shows inhibition constants). However, both α1AT-SMTR/S and α1AT-SLLR/S are more than 10 times stronger inhibitors of PKa than pdC1INH and α1AT-Pittsburgh (Figure 1C). The physiological activity of plasmin is mainly regulated by the SERPIN α2-antiplasmin (inhibition constant k2: 0.96-3.8 × 107 M−1·s−1).41,42 In comparison, pdC1INH inhibits plasmin very modestly (Figure 1D). However, α1AT-SMTR/S and α1AT-SLLR/S, as well as α1AT-Pittsburgh, strongly inhibit plasmin (Figure 1D). The physiological activity of FXIa is regulated by both pdC1INH (Figure 1E; k2: 0.11 × 104 M−1·s−1) and antithrombin (ATIII; k2: 1.7-3 × 102 M−1·s−1).43,44 Interestingly, we found that α1AT-SMTR/S, α1AT-SLLR/S, and α1AT-Pittsburgh are ∼300-fold stronger inhibitors of FXIa than pdC1INH is (Figure 1E). α1AT-WT does not inhibit any of the investigated enzymes in these experiments.
We next investigated the behavior of these α1AT variants in contact system-dependent coagulation assays. For these experiments, we added purified SERPINs (384 nM, or a buffer control, indicated by vehicle) to citrated plasma. In a dilute activated partial thromboplastin time assay (aPTT), α1AT-SMTR/S, α1AT-SLLR/S, as well as α1AT-Pittsburgh, significantly prolongs the clotting time in comparison with vehicle, pdC1INH, and α1AT-WT (Figure 1F). These findings directly correspond to changes in kaolin-triggered thrombin generation (Figure 1G): the variants that prolonged clotting times, delay both the lag time and time to peak of thrombin formation (supplemental Figures 2A and 2B, respectively), as well as the peak height of thrombin formation (supplemental Figure 2C). Only α1AT-Pittsburgh reduces the total amount of thrombin activity that was formed (supplemental Figure 2D), which can be attributed to direct thrombin inhibition by this variant.
The physiological activity of thrombin is mainly regulated by antithrombin (k2: 0.7-7.2 × 103 M−1·s−1).45,46 Heparin enhances the rate of thrombin inhibition by this SERPIN ∼1000-fold (k2: 1.3-7.4 × 107 M−1·s−1).19,46,47 In the absence of heparin, α1AT-Pittsburgh is ∼100 times more potent than antithrombin (k2: 3.1 × 105 M−1·s−1), explains the associated bleeding disorder.24,48 Thrombin favors a proline (P) at the P2 position,49 but we found that it is not essential: a previously published α1AT variant in which P2 is changed from a P to an alanine (A) residue (α1AT-AIAR/S48 ) only displays a modestly reduced capacity to inhibit thrombin (k2: 5.45 × 104 M−1·s−1; supplemental Table 1). Compared with α1AT-Pittsburgh, both α1AT-SMTR/S and α1AT-SLLR/S show strongly reduced thrombin inhibition (Figure 2A). However, α1AT-SMTR/S, similar to α1AT-Pittsburgh, is also a potent inhibitor of FXa (Figure 2B), whereas α1AT-SLLR/S is not. As expected, we found that α1AT-Pittsburgh inhibits clotting in dilute prothrombin time (PT) assays (Figure 2C), as well as in tissue factor (TF)-driven thrombin generation assays (Figure 2D-E; supplemental Figure 3). Both α1AT-SMTR/S and α1AT-SLLR/S prolong clotting to a minor extent at the highest concentrations (Figure 2C). In thrombin generation assays triggered by high TF levels (5 pM; Figure 2D; supplemental Figure 3), we found that only α1AT-Pittsburgh suppresses thrombin formation. However, at low TF levels (0.5 pM; Figure 2E; supplemental Figure 4) both α1AT-SMTR/S and -SLLR/S suppress thrombin generation, while α1AT-Pittsburgh abolishes it.

α1-Antitrypsin variants SMTR/S and SLLR/S target thrombin, FXa, and APC. (A-B,F) Inhibition of enzymes at a fixed SERPIN concentration; table insets show second-order rate constants (k2: 104 M−1·s−1). (A) Inhibition of 17.7 nM thrombin by 38.5 nM SERPIN. (B) Inhibition of 8.5 nM FXa by 192.3 nM SERPIN. (C) Inhibition of dilute PT clotting times in normal plasma in the presence of SERPINs. (D-E) Inhibition of TF-driven thrombin generation in the presence of 384 nM SERPIN. (F) Inhibition of 17.9 nM APC by 96.2 nM SERPIN. (G) Inhibition of APC by 384 nM SERPIN in an aPTT clotting assay. Data are expressed as a ratio of the APC-dependent increase in clotting times. Data represent the mean ± SD of 3 separate experiments, each performed in duplicate. #P < .005; †P < .0005; §P < .0001, compared with pdC1INH by 1-way ANOVA.
α1-Antitrypsin variants SMTR/S and SLLR/S target thrombin, FXa, and APC. (A-B,F) Inhibition of enzymes at a fixed SERPIN concentration; table insets show second-order rate constants (k2: 104 M−1·s−1). (A) Inhibition of 17.7 nM thrombin by 38.5 nM SERPIN. (B) Inhibition of 8.5 nM FXa by 192.3 nM SERPIN. (C) Inhibition of dilute PT clotting times in normal plasma in the presence of SERPINs. (D-E) Inhibition of TF-driven thrombin generation in the presence of 384 nM SERPIN. (F) Inhibition of 17.9 nM APC by 96.2 nM SERPIN. (G) Inhibition of APC by 384 nM SERPIN in an aPTT clotting assay. Data are expressed as a ratio of the APC-dependent increase in clotting times. Data represent the mean ± SD of 3 separate experiments, each performed in duplicate. #P < .005; †P < .0005; §P < .0001, compared with pdC1INH by 1-way ANOVA.
Finally, we investigated the influence of our new α1AT variants on APC activity (Figure 2F). We found that α1AT-SLLR/S has a much lower capacity to inhibit APC than α1AT-Pittsburgh. In contrast, α1AT-SMTR/S still inhibits APC. This is surprising, as this P4-P1 donor RCL sequence (SMTR) originates from FXII, which is not susceptible to APC cleavage (supplemental Figure 1A). We confirmed the activities of the α1AT variants in plasma assays, where both α1AT-SMTR/S and α1AT-Pittsburgh inhibit the anticoagulant effects of APC in plasma coagulation studies (Figure 2G). These data clearly show that both α1AT-SMTR/S and α1AT-SLLR/S have an improved specificity toward the enzymes involved in bradykinin release compared with α1AT-Pittsburgh. Nevertheless, the remaining inhibition of thrombin, FXa, and APC warranted further refinement of the RCL.
Refinement of α1-antitrypsin Pittsburgh’s reactive center loop P1′ residue fully eliminates inhibition of thrombin and APC
We next sought to refine the RCL via mutagenesis of the P1′ amino acid, which is located after the reactive center (Figure 1A). Thrombin favors an alanine (A) or serine (S) at P1′ for optimal substrate conversion.50 Naturally, there is an S present at P1′ in α1AT, and therefore also in the variants that we developed. We hypothesized that replacement with valine (V; naturally present at P1′ in the FXII activation loop), prevents cleavage by thrombin and APC. To examine this hypothesis, we developed 2 novel variants α1AT-SMTR/V and α1AT-SLLR/V.
Both α1AT-SMTR/V and α1AT-SLLR/V no longer inhibit thrombin (Figure 3A, table inset). Furthermore, the change of P1′ residue strongly reduces FXa inhibition by both variants (Figure 3B). As a result, they no longer interfere with coagulation in dilute PT assays (Figure 3C), or thrombin generation at high TF levels (Figure 3D; supplemental Figure 5). However, at low TF concentrations, both variants inhibit thrombin generation (Figure 3E; supplemental Figure 6).
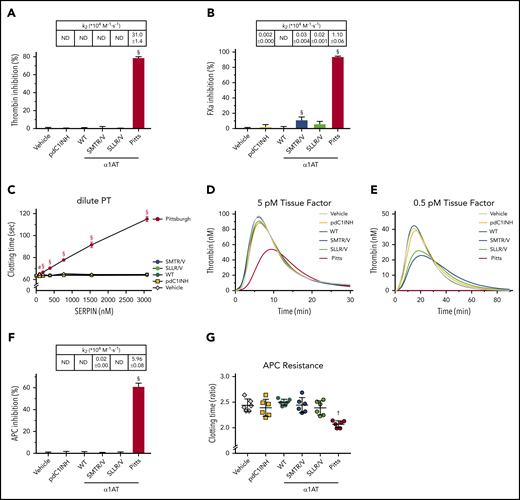
Refinement of α1-antitrypsin Pittsburgh’s reactive center loop P1′ residue fully eliminates inhibition of thrombin and APC. (A-B,F) Inhibition of enzymes at a fixed SERPIN concentration; table insets show second-order rate constants (k2: 104 M−1·s−1). (A) Inhibition of 17.7 nM thrombin by 38.5 nM SERPIN. (B) Inhibition of 8.5 nM FXa by 192.3 nM SERPIN. (C) Inhibition of dilute PT clotting times in normal plasma in the presence of SERPINs. (D-E) Inhibition of TF-driven thrombin generation in the presence of 384 nM SERPIN. (F) Inhibition of 17.9 nM APC by 96.2 nM SERPIN. (G) Inhibition of APC in an aPTT clotting assay by 384 nM SERPIN. Data are expressed as a ratio of the APC-dependent increase in clotting times. Data represent the mean ± SD of 3 separate experiments, each performed in duplicate. #P < .005, †P < .0005, §P < .0001, compared with pdC1INH by 1-way ANOVA.
Refinement of α1-antitrypsin Pittsburgh’s reactive center loop P1′ residue fully eliminates inhibition of thrombin and APC. (A-B,F) Inhibition of enzymes at a fixed SERPIN concentration; table insets show second-order rate constants (k2: 104 M−1·s−1). (A) Inhibition of 17.7 nM thrombin by 38.5 nM SERPIN. (B) Inhibition of 8.5 nM FXa by 192.3 nM SERPIN. (C) Inhibition of dilute PT clotting times in normal plasma in the presence of SERPINs. (D-E) Inhibition of TF-driven thrombin generation in the presence of 384 nM SERPIN. (F) Inhibition of 17.9 nM APC by 96.2 nM SERPIN. (G) Inhibition of APC in an aPTT clotting assay by 384 nM SERPIN. Data are expressed as a ratio of the APC-dependent increase in clotting times. Data represent the mean ± SD of 3 separate experiments, each performed in duplicate. #P < .005, †P < .0005, §P < .0001, compared with pdC1INH by 1-way ANOVA.
The change of P1′ residue eliminated APC inhibition by α1AT-SLLR/V and reduced it by 171-fold for α1AT-SMTR/V (Figure 3F; table inset). As a result, neither variant appreciably inhibits the anticoagulant activity of APC in plasma (Figure 3G). These data show that changing the P1′ residue from S to V reduces off-target interactions. We next investigated whether this change influenced inhibition of enzymes involved in contact activation and bradykinin production.
α1AT-SMTR/V, pdC1INH and α1AT-Pittsburgh inhibit FXIIa with comparable efficacy, whereas α1AT-SLLR/V is ∼twofold weaker (Figure 4A). In contrast, both α1AT-SMTR/V and -SLLR/V show superior inhibition of PKa, with α1AT-SMTR/V being ∼threefold and α1AT-SLLR/V being ∼fivefold stronger than pdC1INH (Figure 4B). α1AT-SMTR/V, -SLLR/V, and pdC1INH inhibit plasmin with similar efficacy but ∼20-fold less strongly than α1AT-Pittsburgh (Figure 4C). Both α1AT-SMTR/V and α1AT-SLLR/V inhibit FXIa much better than pdC1INH (Figure 4D). This is in good agreement with our earlier findings in thrombin generation assays at low TF levels, which together point to FXIa as their primary target (Figure 3E; supplemental Figure 7). Together, these data show that modification of P1′ refines SERPIN specificity, but this appears to come at the price of a drop in efficacy (for an overview, see supplemental Table 1).

α1-Antitrypsin variants SMTR/V and SLLR/V are powerful inhibitors of contact system enzymes. (A-D) Inhibition of enzymes at a fixed SERPIN concentration; table insets show second-order rate constants (k2: 104 M−1·s−1). (A) Inhibition of 25 nM αFXIIa by 96.2 nM SERPIN. (B) Inhibition of 2.3 nM PKa by 28.9 nM SERPIN. (C) Inhibition of 24.1 nM plasmin by 24.1 nM SERPIN. (D) Inhibition of 2.5 nM FXIa by 9.6 nM SERPIN. Effect of 384 nM SERPIN on dilute aPTT clotting times (E) or kaolin-driven thrombin generation (F). Data represent the mean ± SD of 3 separate experiments, each performed in duplicate. #P < .005; §P < .0001, compared with pdC1INH by 1-way ANOVA.
α1-Antitrypsin variants SMTR/V and SLLR/V are powerful inhibitors of contact system enzymes. (A-D) Inhibition of enzymes at a fixed SERPIN concentration; table insets show second-order rate constants (k2: 104 M−1·s−1). (A) Inhibition of 25 nM αFXIIa by 96.2 nM SERPIN. (B) Inhibition of 2.3 nM PKa by 28.9 nM SERPIN. (C) Inhibition of 24.1 nM plasmin by 24.1 nM SERPIN. (D) Inhibition of 2.5 nM FXIa by 9.6 nM SERPIN. Effect of 384 nM SERPIN on dilute aPTT clotting times (E) or kaolin-driven thrombin generation (F). Data represent the mean ± SD of 3 separate experiments, each performed in duplicate. #P < .005; §P < .0001, compared with pdC1INH by 1-way ANOVA.
We next explored the functional properties of α1-antitrypsin variants in contact-system-dependent coagulation assays. Both α1AT-SMTR/V and α1AT-SLLR/V are superior inhibitors compared with pdC1INH in a kaolin-triggered aPTT assay (Figure 4E), as well as in kaolin-triggered thrombin generation assays (Figure 4F). Their presence delays the lag time and time to peak of thrombin formation (supplemental Figures 8A and 8B, respectively), and reduces the peak height of thrombin formed (supplemental Figure 8C). Only α1AT-Pittsburgh was able to diminish the total amount of thrombin formed (supplemental Figure 8D). Overall, these data demonstrate that α1AT-SMTR/V and α1AT-SLLR/V are efficient inhibitors of contact activation. We next set out to test their capacity to inhibit bradykinin formation.
α1-Antitrypsin variants SMTR/V and SLLR/V are powerful inhibitors of bradykinin formation
We first aimed to functionally investigate our α1AT variants in the context of C1INH deficiency. In patients with C1INH-HAE, these levels are well below 50%. As a result, plasma from these patients is intrinsically unstable and prone to preanalytical in vitro contact system activation after blood collection.51 To overcome this technical obstacle, we used a recombinant protein (VAG8) to neutralize C1INH function in plasma.52 Once VAG8 is present in plasma, kaolin-triggered kallikrein-like activity is improperly controlled (supplemental Figure 9A). Under these conditions, α1AT-SMTR/V, α1AT-SLLR/V, and α1AT-Pittsburgh are able to achieve control in a concentration-dependent manner (Figure 5A; we expressed 100% as complete inhibition). By comparison, the C1INH that is naturally present in normal plasma (ie, in the absence of VAG8) inhibits kaolin-triggered kallikrein-like activity by ∼30% (dotted line).
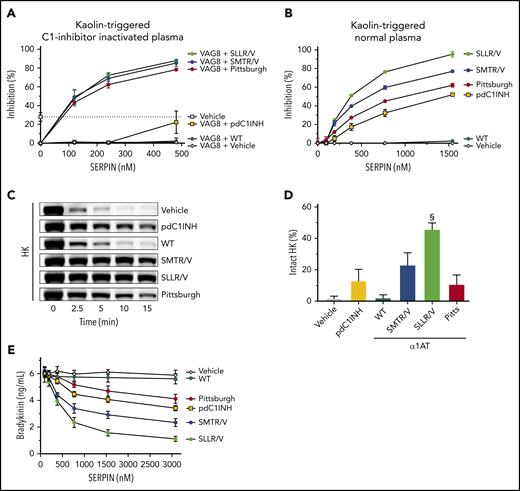
α1-Antitrypsin variants SMTR/V and SLLR/V are powerful inhibitors of bradykinin formation. (A) Inhibition of kaolin-induced contact system enzyme activity by SERPINs in C1INH-inactivated plasma (by 3.37 μM VAG8), measured with a chromogenic substrate. The dotted line indicates the level of inhibition that is achieved by plasma C1INH (ie, in absence of VAG8); 100% indicates full inhibition of enzyme activity. (B) Inhibition of kaolin-induced contact system enzyme activity by SERPINs in normal pooled plasma (with normal C1INH activity), measured with a chromogenic substrate; 100% indicates full inhibition of enzyme activity. (C) Kaolin-induced HK consumption after 15 minutes (quantification of repeated experiments in panel D). (E) Bradykinin formation after 5 minutes in the presence of a concentration range of SERPINs. Data represent the mean ± SD of 3 separate experiments, each performed in duplicate. §P < .0001, compared with pdC1INH by 1-way ANOVA.
α1-Antitrypsin variants SMTR/V and SLLR/V are powerful inhibitors of bradykinin formation. (A) Inhibition of kaolin-induced contact system enzyme activity by SERPINs in C1INH-inactivated plasma (by 3.37 μM VAG8), measured with a chromogenic substrate. The dotted line indicates the level of inhibition that is achieved by plasma C1INH (ie, in absence of VAG8); 100% indicates full inhibition of enzyme activity. (B) Inhibition of kaolin-induced contact system enzyme activity by SERPINs in normal pooled plasma (with normal C1INH activity), measured with a chromogenic substrate; 100% indicates full inhibition of enzyme activity. (C) Kaolin-induced HK consumption after 15 minutes (quantification of repeated experiments in panel D). (E) Bradykinin formation after 5 minutes in the presence of a concentration range of SERPINs. Data represent the mean ± SD of 3 separate experiments, each performed in duplicate. §P < .0001, compared with pdC1INH by 1-way ANOVA.
We next investigated the functional properties of our α1AT variants in the presence of normal C1INH. When pdC1INH (up to 1.4 μM) is added to normal plasma (already contains 2.38 μM C1INH), it modestly inhibits kaolin-triggered kallikrein-like activity in a dose-dependent manner (Figure 5B). Furthermore, it delays HK consumption (Figure 5C-D; supplemental Figure 9B), which reflects bradykinin production. Similarly, pdC1INH modestly suppresses bradykinin release in a dose-dependent manner (Figure 5E) and dampens the burst of bradykinin production (supplemental Figure 9C). By comparison, α1AT-SMTR/V and α1AT-SLLR/V inhibit kaolin-triggered kallikrein-like activity in plasma much better than pdC1INH and even α1AT-Pittsburgh (Figure 5B). In good correspondence, these α1AT variants protect HK from cleavage (Figure 5C-D) and strongly suppress bradykinin release (Figure 5E; supplemental Figure 9C). These experiments point out that α1AT-SLLR/V is a better inhibitor of bradykinin release than α1AT-SMTR/V. This can be attributed to its strong capacity to inhibit PKa (Figure 4B).
Modified SERPINs are antithrombotic in vivo
As our SERPINs block multiple targets in contact system activation,53-55 we next explored whether our α1AT variants have antithrombotic properties. Thrombosis was induced through injury of the carotid artery by FeCl3, and time to vascular occlusion was monitored (Figure 6). We pretreated mice with equimolar amounts of recombinant human C1INH (rC1INH; 16 mg/kg) or α1AT variants (8 mg/kg) by IV tail vein injections. Peak plasma levels of these SERPINs were 1.9 μM (assuming a circulating volume of 2 mL per mouse). Upon challenge, C1INH did not protect against thrombosis (P = .45 vs vehicle). Although α1AT-Pittsburgh showed a trend toward antithrombotic behavior, it failed to reach statistical significance. In contrast, both α1AT-SMTR/V and α1AT-SLLR/V showed significant antithrombotic properties (P = .01 for SMTR/V and P = .04 for SLLR/V vs vehicle, respectively). We attribute the antithrombotic properties of SERPIN variants α1AT-SMTR/V and α1AT-SLLR/V to the multitarget inhibition of contact factors.
![Modified SERPINs are antithrombotic in vivo. Mice (n = 7 per group) were anesthetized, after which the carotid artery was exposed. Mice were pretreated with SERPINs via IV injection (8 mg/kg α1AT variant or 16 mg/kg rC1INH [to correct for SERPIN molecular weights]), after which thrombus formation was initiated by a topical application of FeCl3 (5% wt/vol). Vascular occlusion was monitored by a Doppler flow probe up until 40 minutes. The number of mice with patent carotid arteries after 40 minutes is indicated above the groups. rC1INH, recombinant C1INH. Data are represented as scatterplots with medians. *P < .05, compared with vehicle by Mann-Whitney Student t test.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/134/19/10.1182_blood.2019000481/4/m_bloodbld2019000481f6.png?Expires=1765889340&Signature=u23rowYaziWj4-Aid9L7nLc62GtpzO~f0sQU5WaWIstFclzF-v95NwOJXR173S~pCp9zD4489RHKy4VuDa7~uUicndWbRkHfMOPkbqTrGM592OSVeTyppii0p7JUoho6Z6U0JuP3TXMMe6EUB0tWwUFvH97oSlENnFrRW7S6akj4WqJdXvXi-W~2bnX2N-zBMlIEDiUSbS0PwvCsm5Ze9S8d104OPjvoIz0yyNg8icCwBy~hrZkfXIP1ZO3sG3jprMYYagwTdROfhn-TtdS0TXpXL0buBA0eMoJTE3oVI9QR~fBQ3b6ejbUYqWshCnf9eKes~AswmzOdgA~WsC5iNA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Modified SERPINs are antithrombotic in vivo. Mice (n = 7 per group) were anesthetized, after which the carotid artery was exposed. Mice were pretreated with SERPINs via IV injection (8 mg/kg α1AT variant or 16 mg/kg rC1INH [to correct for SERPIN molecular weights]), after which thrombus formation was initiated by a topical application of FeCl3 (5% wt/vol). Vascular occlusion was monitored by a Doppler flow probe up until 40 minutes. The number of mice with patent carotid arteries after 40 minutes is indicated above the groups. rC1INH, recombinant C1INH. Data are represented as scatterplots with medians. *P < .05, compared with vehicle by Mann-Whitney Student t test.
Modified SERPINs are antithrombotic in vivo. Mice (n = 7 per group) were anesthetized, after which the carotid artery was exposed. Mice were pretreated with SERPINs via IV injection (8 mg/kg α1AT variant or 16 mg/kg rC1INH [to correct for SERPIN molecular weights]), after which thrombus formation was initiated by a topical application of FeCl3 (5% wt/vol). Vascular occlusion was monitored by a Doppler flow probe up until 40 minutes. The number of mice with patent carotid arteries after 40 minutes is indicated above the groups. rC1INH, recombinant C1INH. Data are represented as scatterplots with medians. *P < .05, compared with vehicle by Mann-Whitney Student t test.
Modified SERPINs protect against inflammation in acute and chronic in vivo models of bradykinin-mediated pathology
In mice, carrageenan-induced paw swelling is a model for acute, nonimmune bradykinin-driven inflammatory responses, and it is the gold standard model for development of therapeutic agents against HAE.56,57
On carrageenan injection (n = 12 mice per group), paw size rapidly increases to ∼150% within the first 10 minutes (Figure 7A; supplemental Figure 10A). Hereafter, the swelling reaction progresses steadily (Figure 7A; supplemental Figure 10B-D). This inflammatory reaction is almost completely abolished (115% vs 150%) when mice are IV pretreated with indomethacin (5 mg/kg), a nonsteroidal anti-inflammatory cyclooxygenase inhibitor.58 Pretreatment with the bradykinin B2-receptor antagonist icatibant (1 mg/kg) or rC1INH (16 mg/kg) significantly reduces swelling after 10 minutes (138% or 125% vs 150%, respectively; Figure 7A; supplemental Figure 10). In this model, we found that pretreatment with modified α1AT variants (8 mg/kg) confers protective effects: α1AT-WT, α1AT-SMTR/V, α1AT-SLLR/V, and α1AT-Pittsburgh all significantly reduced paw swelling (Figure 7A; supplemental Figure 10). The protective effect of α1AT-WT is explained by its natural capacity to inhibit neutrophil elastase, which is implicated in this model.59 Similarly, the inhibition of thrombin by α1AT-Pittsburgh can lead to additional protection on top of its ability to inhibit the formation of bradykinin.60 Nevertheless, when directly compared with α1AT-WT, α1AT-SMTR/V, α1AT-SLLR/V, and α1AT-Pittsburgh were significantly better at preventing paw swelling after 6 hours (supplemental Table 6).

Modified SERPINs protect against inflammation in acute and chronic in vivo models of bradykinin-mediated pathology. (A) Carrageenan-induced paw swelling. Mice (n = 12 per group) were pretreated by IV injection (8 mg/kg α1AT variant, 16 mg/kg rC1INH, 1 mg/kg icatibant (HOE140) or 5 mg/kg Indomethacin). Hereafter (T = 0), mice were challenged by carrageenan injection into the dorsal side of the left hind paw. Changes in paw size were measured in triplicate before and during the carrageenan challenge. Data represent the mean ± SD. (B) DSS-induced colitis. On day 0 and 3, mice (n = 6 per group) received an IV injection with inhibitor (8 mg/kg α1AT variant, 16 mg/kg rC1INH). Hereafter (T = 0), mice were challenged with DSS (3% wt/vol) via their drinking water. The control group (baseline) received no DSS. After 6 days, mice underwent a gavage with FITC-dextran to determine epithelial leakage of the gut. Four hours after the gavage, mice were euthanized and FITC-DXS plasma levels were determined. Data are represented as box and whiskers plot (5% to 95% percentile). #P < .005, compared with vehicle by 1-way ANOVA with post-hoc Dunnett’s multiple comparison test.
Modified SERPINs protect against inflammation in acute and chronic in vivo models of bradykinin-mediated pathology. (A) Carrageenan-induced paw swelling. Mice (n = 12 per group) were pretreated by IV injection (8 mg/kg α1AT variant, 16 mg/kg rC1INH, 1 mg/kg icatibant (HOE140) or 5 mg/kg Indomethacin). Hereafter (T = 0), mice were challenged by carrageenan injection into the dorsal side of the left hind paw. Changes in paw size were measured in triplicate before and during the carrageenan challenge. Data represent the mean ± SD. (B) DSS-induced colitis. On day 0 and 3, mice (n = 6 per group) received an IV injection with inhibitor (8 mg/kg α1AT variant, 16 mg/kg rC1INH). Hereafter (T = 0), mice were challenged with DSS (3% wt/vol) via their drinking water. The control group (baseline) received no DSS. After 6 days, mice underwent a gavage with FITC-dextran to determine epithelial leakage of the gut. Four hours after the gavage, mice were euthanized and FITC-DXS plasma levels were determined. Data are represented as box and whiskers plot (5% to 95% percentile). #P < .005, compared with vehicle by 1-way ANOVA with post-hoc Dunnett’s multiple comparison test.
Next, we set out to test our α1AT variants in another, more chronic model of bradykinin-driven pathology. Ulcerative colitis is a chronic inflammatory bowel disease with relapsing attacks. In mice, important features of colitis can be induced via dextran sulfate sodium (DSS; 3% wt/vol) administration in drinking water during a 6-day period.61,62 HK and PK deficiency are protective in this mouse model,15 and icatibant treatment has therapeutic effects.63 On days 0 and 3, we IV administered α1AT variants or rC1INH (8 mg/kg and 16 mg/kg, respectively) to mice (n = 6 per group). As previously described, mice began to experience loss of bodyweight after day 5 (supplemental Figure 11) in comparison with unchallenged control mice (No DSS; baseline). In this timeframe, we observed no differences between groups: all challenged groups had lost ∼10% bodyweight.
However, a perturbed epithelial permeability is a hallmark feature of colitis. To investigate this phenomenon, mice were given a gavage with a fluorescent tracer (FITC-dextran) after the challenge (on day 6). Four hours later, mice were exsanguinated to determine FITC-dextran levels in their plasma. As expected, the DSS challenge causes plasma levels of FITC-dextran to increase, indicating an increase in epithelial permeability (Figure 7B; unchallenged vs vehicle P = .002). Pretreatment with rC1INH, α1AT-WT, α1AT-SMTR/V, or α1AT-Pittsburgh did not significantly lower plasma FITC-dextran levels. However, pretreatment with α1AT-SLLR/V caused a significant reduction in plasma FITC-dextran levels (P = .0097 vs vehicle). This shows that our most powerful PKa-blocking α1AT variant (Figure 5) is protective against pathogenic epithelial leakage.
Discussion
Bradykinin is a powerful mediator of inflammation. Its production is dependent on a balanced interplay between contact system enzymes, C1INH, and bradykinin metabolism. In this study, we redesigned the broad-spectrum SERPIN α1AT-Pittsburgh (P4-P1′ RCL: AIPR/S) for selective inhibition of contact activation and blockade of bradykinin production. Our initial P4-P1′ variants (SMTR/S and SLLR/S) showed increased target specificity over α1AT-Pittsburgh (Figure 1), but still displayed residual reactivity with thrombin, FXa, and APC (Figure 2). For further refinement, we subsequently replaced the P1′ residue of both variants by a valine (V), which strongly lowers their ability to inhibit FXa and eliminates their ability to inhibit thrombin and APC (Figure 3). This increase in specificity comes at the price of a lowered ability to inhibit PKa and plasmin (supplemental Table 1). Nonetheless, α1AT-SMTR/V and α1AT-SLLR/V are superior to C1INH as inhibitors of kaolin-triggered bradykinin production in both normal plasma and C1INH-inactivated plasma (Figure 5).
The contact system has been repeatedly implicated to contribute to thrombosis in mouse models.53-55 We found that pretreatment of mice with SERPIN variants α1AT-SMTR/V and α1AT-SLLR/V conferred protection against arterial thrombosis after FeCl3-induced injury of the carotid artery (Figure 6). Administration of rC1INH was ineffective, showing that the inhibitory capacity of endogenous C1INH is unable to withstand contact activation and cannot be rescued by providing more of the same inhibitor. Although treatment with α1AT-Pittsburgh showed a trend toward antithrombotic behavior in our experiments, it did not reach statistical significance. This may be attributable to experimental variability or to its promiscuous capacity to simultaneously block pro- and anticoagulant enzymes.19,21
In an in vivo model of acute bradykinin-driven inflammation, both α1AT-SMTR/V and SLLR/V block tissue swelling after topical injections with carrageenan (Figure 7A). Although this model is not a specific disease model for HAE (ie, it is not a model for C1INH-deficiency), it is generally applied for HAE therapy development56
Although bradykinin is the main disease mediator in HAE, it is also implicated in allergic reactions,64 brain edema,65,66 asthma,67 arthritis,16 colitis,15 and sepsis.68 In an in vivo model for colitis, we found no improvement in bodyweight when mice were treated with SERPINs (supplemental Figure 11). This contradicts earlier studies in which extensive administration (0.3-1.5 mg/kg subcutaneous twice daily) of icatibant protected against weight loss in the same model.63 It is possible that an alternative treatment regimen might influence our findings. Excitingly, we found that α1AT-SLLR/V, our most powerful inhibitor of PKa, protected mice against DSS-induced epithelial leakage (Figure 7B). In this same model, α1AT-SMTR/V was ineffective. We attribute this to its lowered ability to inhibit bradykinin production (Figure 5). However, it is noteworthy that this SERPIN very weakly inhibits APC under conditions using purified components (Figure 3F), although it does not affect the anticoagulant function of APC in plasma. Nonetheless, we currently cannot rule out that this SERPIN variant interferes with the natural cytoprotective role of APC in this setting.69
The application of designer SERPINs reaches beyond bradykinin-driven disease. During the past years, α1AT has been adapted to inhibit enzymes such as furin70-72 and complement factor C1s.73 Despite their success in changing the specificity, in vivo proof-of-concept studies are scarce. Furthermore, multiple studies investigated the therapeutic value of α1AT-Pittsburgh for sepsis.20,74 Very recently, the α1AT P2-P1′ variant KR/K (α1AT-KR/K) was identified as a specific inhibitor of APC.24 Here the authors showed that selective inhibition of APC was able to rescue thrombin generation in both hemophilia type A and B plasma. When FIX-deficient mice were pretreated with 7.5 mg/kg α1AT-KR/K, they were protected in a tail-clip bleeding model. Although we and others demonstrated the efficacy of modified SERPINs through IV pretreatment, there are limitations to the recurrent administration of recombinant proteins for prophylactic treatment of chronic diseases. Fortunately, there are exciting developments in the gene-therapy field. Using adenovirus-based gene-therapy, α1AT can be stably expressed at meaningful levels in mice for up to 24 weeks.75 This approach is currently undergoing phase 1/2 trials for the treatment of α1AT deficiency in human subjects.76 This might ultimately pave the way for gene-therapy-based strategies to employ designer SERPINs for the treatment of chronic diseases.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Dan Murphy for critically reading the manuscript and Jasper Kers for valuable technical assistance.
C.M. gratefully acknowledges the Landsteiner Foundation for Blood Transfusion Research (#LSBR 1520), the Netherlands Thrombosis Foundation (#201703), and the Netherlands Organization for Scientific Research (NWO, #TTW:00670785). T.R. acknowledges the German Research Foundation (#A11/SFB 877 and B8/SFB 841) and a European Research Council grant (F-12). W.S. gratefully acknowledges financial support from the Royal Thai Government. The in vivo proof-of-concept studies were financially supported by SERPINx, a biotech spinout company of University Medical Center Utrecht.
Authorship
Contribution: S.d.M., W.S., R.K.M., M.H., J.C.M.M., G.P., T.R., and C.M. conceived and/or designed the study; S.d.M., W.S., R.M.K., and N.M.J.P. performed in vitro experiments; R.K.M. performed the in vivo carotid occlusion model; and S.d.M., W.S., and C.M. wrote the manuscript.
Conflict-of-interest disclosure: C.M. is consultant to Shire. C.M., M.H., and S.d.M. are founders of SERPINx BV, a biotech spinout company of University Medical Center Utrecht (to develop α1AT-SMTR/V and SLLR/V). C.M. and S.d.M. participate in revenue sharing through the commercialization arm of the University Medical Center Utrecht. The results discussed in this manuscript form part of the patent application, "Modified serpins for the treatment of bradykinin-mediated disease" (WO 2018/154044 A1) (C.M. and S.d.M.). The remaining authors declare no competing financial interests.
Correspondence: Coen Maas, University Medical Center Utrecht, Heidelberglaan 100, 3584 CX Utrecht, The Netherlands; e-mail: cmaas4@umcutrecht.nl.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal