


Abstract
Chronic myeloid leukemia (CML) is the model cancer, demonstrating the clinical benefits of targeted therapy and the power of molecular diagnostics and monitoring. In CML, the BCR-ABL1 fusion gene and its companion messenger RNA offers a unique target differentiating cancer from the normal cell, affording the potential for very sensitive and specific assays. Because CML is such an ideal model, new methods are arising that should make testing in CML faster, more reliable, and reach a greater sensitivity. New ultrasensitive sequencing approaches, coupled with single-cell genomic approaches, further the study of measurable residual disease, clonal heterogeneity, and promise to make clinical trials more innovative and informative. These methods should be able to be transferred to other hematological and solid malignancies.
Introduction
Say you were assigned to create, from scratch, a model system to study cancer. What would be the desired features of your model? To study initiation, you would want a fairly simple genetic event (maybe a translocation?). Because cancer routinely progresses and metastasizes, you would want a model in which progression is the rule. It would also be important to be able to track the disease with great sensitivity and sensitivity, and of course, have a drug that targets the specific genetic lesion that drives the disease so you can understand what a cure looks like.
Behold, chronic myeloid leukemia (CML).
CML as a model of targeted therapy and disease monitoring
CML is the “poster child” of genetically informed therapy because the unique chimeric BCR-ABL1 DNA, RNA, and protein is the target for tyrosine kinase inhibition and disease monitoring. In this paper, we will use CML to describe future diagnostic assays, the questions these assays can help answer, and the new questions that can now be considered given the new methodology.
First, a few basic facts on CML in the age of tyrosine kinase inhibitor (TKI) therapy.
TKI therapy has changed the natural history survival of chronic phase CML patients from 5 to 7 years to now a lifespan that approximates the general population. Indeed, ∼90% of patients treated with imatinib in the International Randomised Study of Interferon versus STI571 (IRIS) trial were still alive after 6 years of follow-up.1,2 Rates of response may be even higher with second-generation TKIs compared with imatinib, although thus far, an overall survival advantage has not been clearly demonstrated compared with imatinib.3-7
Peripheral blood BCR-ABL1 messenger RNA (mRNA) levels provide a fast, accurate, and clinically useful indicator of disease activity and burden. Moreover, BCR-ABL1 levels at specific time points are highly correlated with outcome measurements. Achievement of a major molecular response (MMR; corresponding to a 3 log reduction and BCR-ABL1 ≤0.1% on the International Scale) puts the patient in a “safe harbor” where resistance and progression are unlikely.8,9 Furthermore, the demonstration of a “deep molecular response” (MR4 or lower) can identify patients who may successfully discontinue TKI therapy (preferably on a clinical trial).10-12
However, resistance is not futile. Resistance is categorized as primary (poor initial response to therapy) or secondary (resistance after initial response to TKI therapy). In the latter category, point mutations in the ABL tyrosine kinase domain that influence TKI binding are detected in approximately 1/3 of patients with TKI resistance.13,14
In cases of resistance, changing therapy to another TKI yields at least a cytogenetic remission in ∼50% of the time.15 However, patients who respond to second-line salvage therapy often relapse, often with a new mutation.16
Approximately 50% of patients with “deep” molecular responses (as defined previously) are able to discontinue their TKI therapy and stay off therapy without a molecular relapse (treatment-free remission).17 Those patients who relapse generally do so with a few months of discontinuation, so frequent monitoring post-TKI cessation is warranted.
Methods of CML monitoring
To better appreciate the motivation to create better technologies to diagnose and monitor CML, we should first consider the current methods at our disposal.
The t(9,22) reciprocal translocation is the basis of pathology, diagnosis, and monitoring in CML. In the vast majority of cases, BCR-ABL1 transcripts arise by the fusion of exons 13 or 14 of the BCR gene to exon 2 of the ABL gene (e13a2 and e14a2, respectively), thus coding for the 210-kDa protein product.18 In Ph+ acute lymphoblastic leukemia, the major isoform results from the fusion of exon 1 of the BCR gene with exon 2 of the ABL gene (e1a2), yielding a 190-kDa protein product.18,19
The Ph, BCR-ABL1 DNA, and BCR-ABL1 mRNA serve as “fingerprints” that clearly differentiate the leukemia clone from normal cells. Methods to detect these markers are shown in Table 1. Metaphase cytogenetics detect the Ph as well as any other chromosomal abnormalities, yet is functionally limited because most tests look at a limited number of metaphases (generally, twenty). Fluorescence in situhybridization is more sensitive than cytogenetics in detecting the BCR-ABL1 fusion and can be performed on bone marrow or peripheral blood, yet will not detect additional chromosomal changes unless specific probes to those areas are used.
Tests now and in the future
| Purpose | Test now | Test soon | Faster? | Cheaper? | Better? |
|---|---|---|---|---|---|
| Detection of Ph | Cytogenetics, fluorescence in situ hybridization | In situ BCR-ABL1 protein assay | Yes | No | Quantitation of protein targets could be clinically useful |
| BCR-ABL1 mRNA detection | qRT-PCR | Digital PCR | Yes | No | Potentially more accurate, sensitive, and robust |
| BCR-ABL1 DNA detection | Multiplex PCR and sequencing | NGS sequencing or real-time sequencing | Yes | No | Could simplify BCR-ABL1 DNA testing |
| ABL TKD mutation testing | Sanger or NGS | Ultrasensitive NGS | No | No | Great sensitivity, but unclear which clinical settings will benefit |
| Diagnosis and monitoring in low-income areas | Some local, but more outsourced testing | Dried blood spots or point-of-care devices | Yes | Yes | Potentially gets the test to more patients. Lack of sensitivity compared with standard PCR |
| Purpose | Test now | Test soon | Faster? | Cheaper? | Better? |
|---|---|---|---|---|---|
| Detection of Ph | Cytogenetics, fluorescence in situ hybridization | In situ BCR-ABL1 protein assay | Yes | No | Quantitation of protein targets could be clinically useful |
| BCR-ABL1 mRNA detection | qRT-PCR | Digital PCR | Yes | No | Potentially more accurate, sensitive, and robust |
| BCR-ABL1 DNA detection | Multiplex PCR and sequencing | NGS sequencing or real-time sequencing | Yes | No | Could simplify BCR-ABL1 DNA testing |
| ABL TKD mutation testing | Sanger or NGS | Ultrasensitive NGS | No | No | Great sensitivity, but unclear which clinical settings will benefit |
| Diagnosis and monitoring in low-income areas | Some local, but more outsourced testing | Dried blood spots or point-of-care devices | Yes | Yes | Potentially gets the test to more patients. Lack of sensitivity compared with standard PCR |
Each future test is assayed against the faster/cheaper/better criteria.
Quantitative reverse transcriptase polymerase chain reaction (qRT-PCR) of BCR-ABL1 mRNA is the most sensitive method for monitoring CML. In most hands, the assay can detect CR-ABL1 transcript in a background of ∼100 000 total mRNA transcripts.20 CML disease burden is monitored by the reduction in BCR-ABL1 transcripts from a standardized value (the International Scale). The amount of disease burden reduction is denoted by molecular response (MR) followed by the log amount of reduction. Thus, MR2 refers to a 2-log reduction on the International Scale. The concept of an MMR was initially defined as a ≥3-log reduction in BCR-ABL1/BCR in the IRIS trial and became an established response metric because achievement of MMR correlates favorably with long-term outcomes.9 This 3-log reduction (MR3) is thus 0.1%IS.
Mutation detection
Mutations in the ABL kinase domain are in important part of TKI resistance, occurring in roughly 25% to 50% of cases of secondary resistance.21 The acquisition of these mutations and the loss of response are associated with a heightened risk of progression to advanced-phase disease. The detection of mutations is limited by the sensitivity of available assays. The most common method of direct (“Sanger”) nucleotide sequencing can detect an ABL tyrosine kinase domain mutation if the mutated clone composes 10% to 20% of the total sample.9 The low sensitivity of Sanger sequencing limits its use to identify early point mutations before frank resistance. Thus, a rising BCR-ABL1 level, especially toward or greater than MMR, suggests a group worthy of testing for an ABL mutation.22
Searching for better mousetraps
“Fast, Cheap, and Good… pick two. If it’s fast and cheap it won’t be good. If it’s cheap and good it won’t be fast. If it’s fast and good it won’t be cheap. Fast, cheap and good… pick two words to live by.”
—jim jarmusch, as told to tom waits
It does not take much motivation to create faster, smaller, better versions of technologies (it’s what we geeks do). In the case of CML, advances are focused on the 3 most pressing issues in advancing patient care: (1) more sensitive assays that may make predicting and monitoring attempts at TKI discontinuation more successful; (2) better tests to detect resistance mutations, which may allow for the early change of therapy to prevent resistance and progression; and (3) methods that can be used to advance our knowledge about the basic biology of CML and its course following therapy.
Methods to increase the sensitivity of minimal residual disease detection
The need for more sensitive BCR-ABL1 detection is driven by the new interest in TKI discontinuation for those with very low (MR ≤4.0) BCR-ABL1. For cases without detectable BCR-ABL1, can methods be developed that can further assess disease burden? Will these levels or residual disease predict cases that can successfully discontinue therapy or those who will relapse?
Digital PCR.
The concept of signal to noise explains the difficulty of detecting rare targets (the signal) in a vast background of nontarget (the noise). “Digital” PCR is a methodological approach to beating the signal/noise limitation.23 The concept is to divide the sample into an excess of partitions, so that each partition houses a single copy of either the target (signal) or the background (noise). Amplification yields a positive signal with target amplification (digital code = 1), and no amplification in partitions without the target (= 0). The Poisson distribution is then used to estimate the initial preamplification BCR-ABL1 copy number. Some publications suggest that digital PCR increases the limit of detection of BCR-ABL1 mRNA by >1 log.24 The method has the additional advantages of having low technical variation, and calibration curves are not required to yield an absolute numerical value.25 Currently, there are 2 strategies for partitioning either by pumping template through microfluidic tubes into miniscule wells or mixing PCR reagents and template into machine-generated bubbles. Both technologies can be used for ultrasensitive detection of BCR-ABL1 or specific ABL mutations.26
DNA PCR.
Amplification of BCR-ABL1 DNA instead of RNA has several potential advantages. DNA is inherently more stable than RNA, which is potentially important when samples must be shipped to testing sites. In addition, the number of BCR-ABL1 molecules vary from patient to patient (or within a patient, from cell to cell), whereas the number of BCR-ABL1 DNA copies per cell is more predictable. In addition, an assay using DNA can be standardized to a cell number using a DNA control gene as a surrogate. The difficulty of using BCR-ABL1 DNA is that the BCR and ABL breakpoint regions are very large. Thus, multiple sets of primers must be used to first find the patient’s particular breakpoint. Once the BCR-ABL1 DNA breakpoint is sequenced, specific primers can be generated and used in subsequent assays (much akin to IgH V-D-J PCR quantification). The DNA approach can have a level of sensitivity down to 1:106 in many cases, which is greater than in qRT-PCR. There appears to be a good correlation between DNA qPCR and qRT-PCR, with most discordance on the side of DNA detection in the absence of RNA detection. Remarkably, in these cases of RNA negativity, the DNA test detects minimal residual disease (MRD) in nearly 50% of cases.27
The amplification and detection of BCR-ABL1 might be streamlined with assay development copying the next-generation sequencing (NGS) methods used to monitor disease burden in acute lymphoblastic leukemias, in which high throughput sequencing of the complementarity determining region 3 regions of IGH or T-cell receptor genes, TRB and TRG, can provide unprecedented sensitivity (exceeding 10−5) and increased specificity for tracking leukemia-specific clonal gene rearrangements.28-30 The secret was the design of multiple “forward” and “reverse” primers, carefully selecting primer design so that no amplification bias could occur for any 2 sets of primers. Similarly, one can imagine multiple BCR and ABL primers spanning large genomic expanses, with selection of the primers in which amplification across the breakpoint is permitted.
Methods to increase availability of diagnostic and MRD testing
In the Western Educated Industrialized Rich Democracies31 (WEIRD) world, the diagnosis of CML is routine, but the sticking point is adherence to the suggested BCR-ABL1 monitoring schedule thought critical to optimal treatment and outcomes. Easier and faster BCR-ABL1 testing that can be expanded to more locations might be expected to improve testing adherence. However, in areas of low resources, the barriers in CML encompass access to diagnostic testing, TKI availability, and monitoring. The obstacles include basic needs such as traveling to clinics, availability of needles, blood tubes, and electricity. These daunting obstacles need to be overcome if access to curative therapies is to be provided to most of the world’s cases of CML.
“Automated” PCR.
The advent of automated PCR molecular testing using prepackaged cartridges brings the advantages of less operator participation (and thus, error), increased efficiency (no need to batch), and greater speed to test result.32 There are further advantages in controlling amplimer cross contamination and rapid (same day) result turnaround, very positive features in cases where patients must travel long distances to undergo testing. The availability of the Cepheid BCR-ABL1 test was greatly accelerated by the World Health Organization’s enthusiasm toward Cepheid’s infectious disease assays (eg, tuberculosis, HIV), which has led to machines placed in many regional centers. Because the box is agnostic to what is being assayed, this allows CML patients to potentially to run alongside of cases testing for other diseases.
Isothermal PCR.
The dominant technology to perform nucleic acid amplification is PCR, but this requires thermocycling, and the application of this energy means electricity. This is an obvious pitfall for much of the world. Isothermal amplification occurs at a single temperature, resulting in the potential to amplify target sequences without electricity, making it attractive for point-of-care devices in areas of low resources.33 Two isothermal methods have been used for BCR-ABL1 fusion transcript detection, nuclei acid sequence-based amplification34 and loop-mediated isothermal amplification (LAMP). The former works by using reverse transcriptase to create a double-stranded DNA molecule, followed by T7 RNA polymerase for complementary RNA amplification. LAMP uses the Bst polymerase enzyme and specialized primers to promote strand displacement and high replication activity, producing such a large amount DNA product that it can be detected by ultraviolet light or DNA stains. Studies using LAMP to detect the PML-RARA transcript in acute promyelocytic leukemia reliably demonstrate isothermal PCR’s feasibility for rapid and simple diagnostics.35
Dried blood spot BCR-ABL1 detection.
Another alternative for BCR-ABL1 testing in areas of low resources is to send samples to specialized centers. The major disadvantage is the probative cost of sending a blood sample by international mail, which costs hundreds of dollars per shipment. Moreover, unless many patients visit the clinic in a very short span, costs cannot be decreased by batching. An attractive option is dried blood spots. DNA is fairly stable on commercially available paper, whereas RNA is not. However, it appears that although RNA in paper degrades, enough persists after weeks of snail mail that an accurate BCR-ABL1 measurement can be obtained.36 Moreover, samples from several days can be batched, further driving down costs. RNA and DNA can be obtained from the same paper, allowing for BCR-ABL1 quantification, and if clinically indicated, ABL mutation testing.
Methods for BCR-ABL1 resistance mutations
The sooner a mutation can be detected, the earlier alternative therapy can be started to curb the emergence of the resistant clone. This is of particular interest given the close relationship of resistance and progression to advanced phase disease.37,38
More sensitive sequencing.
As noted, the limit of sensitivity of Sanger sequencing is ∼10% to 20% (mutation to wild-type frequency) and has limited use in early detection of mutations or following MRD. NGS has a better sensitivity and has been shown to be effective in earlier detection of mutation, though random errors generated by NGS library preparation (particularly when starting from mRNA) limit the mutation detection sensitivity to ∼1%.39,40 Novel “error-correcting” approaches have been developed or applied to address these challenges, including bar-coding through use of single-molecule molecular inversion probe tagging and duplex sequencing.41,42 In duplex sequencing, both DNA strands are simultaneously sequenced. True mutations are called only when the correct complementary base changes are found in both strands. In contrast, random mutations will be only in 1 strand, but the complementary mutation will not be found in the opposite strand.43,44 This theoretically makes a method induced false-positive mutation quite unlikely (the technical error rate is ∼0.001%). Figure 1 shows how duplex sequencing leads to unambiguous mutation calling in BCR-ABL1 with a kinase domain mutation that is hidden in the background of NGS. Although we have demonstrated the power of duplex sequencing to detect rare BCR-ABL1 mutations, a real boom would be its use to follow MRD in diseases characterized by a limited number of mutations, such as acute myeloid leukemia (AML). This methodology may be very important in defining intratumoral heterogeneity at currently undetectable levels, and as MRD detection, where it has the potential to be several orders of magnitude more sensitive than flow cytometry.
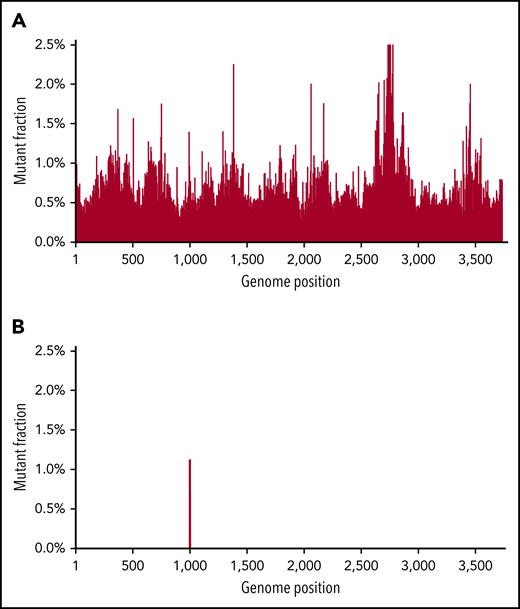
An example of duplex sequencing of a T315I mutation. Duplex sequencing eliminates background “noise” found in NGS. Top, sequencing of a CML patient with a known T315I ABL tyrosine kinase domain mutation. NGS found background mutations in almost all base pair regions at the 1% level. Bottom, with duplex sequencing, the background technical artifacts are eliminated, with only the true mutation remaining. Figure reprinted with courtesy from Schmitt et al.59
An example of duplex sequencing of a T315I mutation. Duplex sequencing eliminates background “noise” found in NGS. Top, sequencing of a CML patient with a known T315I ABL tyrosine kinase domain mutation. NGS found background mutations in almost all base pair regions at the 1% level. Bottom, with duplex sequencing, the background technical artifacts are eliminated, with only the true mutation remaining. Figure reprinted with courtesy from Schmitt et al.59
Real-time sequencing.
Two systems are available that can yield real-time nucleic acid sequencing. One uses a platform with an ultrasensitive metallic light detection system attached to the bottom of glass wells, where a polymerase is attached. When a DNA template is present, fluorescent-labeled phosphor-linked nucleotides are added into the newly extending DNA strand, and a light pulse results in an excited fluorochrome that emits a momentary burst of colored light read by the sensor. The nucleotide-specific light pulses thus provide a real-time DNA sequence.45,46 A second system uses a synthetic polymer membrane studded with a DNA helicase nanopore protein. This pore enzymatic shows activity at 1 opening and a small internal passage chamber that limits excursion to single nucleotide base size. As the DNA molecule courses through the nanopore, individual nucleotide bases emit small, measurable changes in the electrical field of the synthetic polymer membrane. Remarkably, specific nucleotide bases can be identified by the electrical field change.47 The speed and range of sequencing is striking, as kilo-megabase DNA reads are possible at speeds of hundreds of base pairs per second. These techniques may prove useful in several facets of CML diagnostics and monitoring. First, the ability to make huge sequencing reads makes it a potential platform to efficiently fingerprint the BCR and ABL DNA translocation, making it relatively easy to identify regions for patient-specific primers for use in DNA monitoring. These platforms may be multiplexed to include simultaneous detection of unique fusions in other disease that might drive CML biology.48
Methods to understand the biology of CML
Despite our great progress in CML, we do not understand why some patient have a poor initial response to therapy, why some patients can obtain a deep response, why some with a deep response can have their TKI discontinued and stay in remission, or why patients become resistant and progress to blast crisis. The usual methods (DNA sequencing of the usual suspects, RNA seq., etc.) are being used to address these problems. What new approaches might be useful in understanding the biological of CML initiation, response, and resistance/progression?
In situ flow/RT-PCR.
The measure of disease burden by RNA or DNA is proximal to the ultimate outcome of the Ph, the BCR-ABL1 protein. There is the potential that the best measure tying disease burden and disease behavior will be at the level of quantification of the BCR-ABL1 protein. Flow cytometry-based assays have been developed to capture, label, and detect the BCR-ABL1 fusion protein. Work thus far suggests the flow detection of BCR-ABL1 protein has lower specificity compared with conventional cytogenetic or PCR assays of BCR-ABL1, but similar positive/negative predictive value and sensitivity.49
Single-cell analysis of gene mutations and expression.
Most genetic analysis, be it genotyping, sequencing, gene expression, or methylation, is based on analysis of bulk material; thus, the results represent an average of the complexity of the cancer. Early studies trying to get at clonal complexity used colonies derived from single cells simply because of the technical ease of doing analyses on hundreds/thousands of cells rather than 1. This technique is potentially very biased because assays were only performed cells that could form colonies, and these may be a rarity not reflecting the biology of the actual cancer. A cleaner approach is analyzing single cells isolated by flow cytometry, but this is painfully exacting, very expensive, and not scalable.
Several techniques use droplets as reaction chambers have emerged. The principles are similar: cells are fused into droplets containing bar-coded primers that can be used to either sequence RNA (eg, 10X Genomics) or DNA targets (eg, Mission Bio). The material from all the cells are combined and sequenced. The readout of the barcodes identifies RNA/DNA and cell (“I am the Ras gene mRNA from cell #3”). For RNA, this allows the types of cells in the sample (eg, T cells, B cell, monocytes), their numbers, and what they are doing (inferred by the RNA expression in the particular cell) to be seen. This gives a unique picture of the cancer “ecosystem.” In addition, single-nucleotide polymorphisms can be potentially identified, which can be quite useful in understanding relapse in the allogeneic transplant setting.50
Single-cell RNA may be a method to understand resistance in CML.51 The study of distinct cell populations based on multiple leukemia stem cell markers has been performed on CML diagnostic and treated samples. Single-cell mRNA on Lin−CD34+CD38−/low cells inferred a heterogeneous leukemia stem cell population at diagnosis that persisted in TKI-resistant cases. Moreover, single-cell expression was able to demonstrate the change in cell subsets of a total population (the ecosystem), including different types of cell lineages and differentiation states within lineages.
Single-cell DNA genotyping potentially offers a unique insight into intratumoral heterogeneity and how it changes with the selective pressure of treatment. Much of this work is in AML, but could readily be applied in CML. Single-cell genotyping in AML suggests multiple clones at diagnosis, with the usual pattern of clonal dominance of a few clones and several small subclones.52 Because gene–gene interactions are critically important for determining the leukemic potential of residual cells, understanding if mutations occur with the same cells is likely important to know. The pattern of resistance to cancer therapy suggests Darwinian selection, and thus, single-cell approaches to look at the dynamics of clonal structure, competition, cooperation, and selection is needed to understand the cancer ecosystem.
Conclusion: with new tools, new questions and problems
The common point of debate in laboratories, grant reviews, coffee shops, and (if grant reviews do not go well) bars is that work that centers on techniques, methods, and devices is “descriptive” and is the sad and ugly stepchild to “mechanistic” research. This is simplistic. New methods not only allow us to investigate known questions, but also reveal new questions we had not even considered. New technologies create a new window to the world.
Table 1 and Figure 2 outlines the tests currently used in CML and how the new methods described here will improve our clinical skills and biological understanding. Beyond CML, and even beyond leukemia, one can imagine that these more sensitive, more comprehensive techniques will help us understand what it takes to cure cancer.
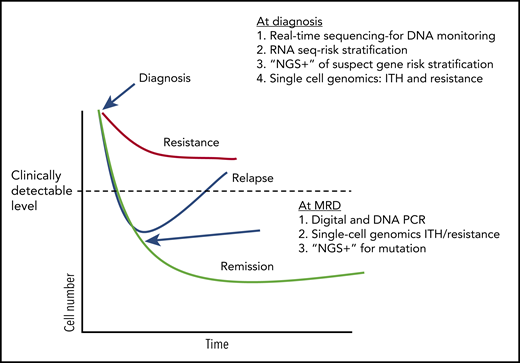
Treatment response and potential uses of emerging technologies. A conjecture on how tests may be used in the future to guide CML (and other hematological malignancies) therapy, based on current research work in the field (ignoring any financial considerations). At diagnosis, DNA methods can be used to both quickly determine breakpoints useful in designing DNA monitoring assays and other mutations that might influence initial response to TKI therapy. RNA sequencing (RNA seq) can used to assay specific genes and pathways associated with early response. Single-cell genetics can be used to establish potentially troublesome complex heterogeneity and identify populations with a resistance signature. During therapy, digital and DNA PCR can be used as a more sensitive determinant of deep molecular response, and “NGS+” used to identify mutations in patients without a deep response who appear to be relapsing.
Treatment response and potential uses of emerging technologies. A conjecture on how tests may be used in the future to guide CML (and other hematological malignancies) therapy, based on current research work in the field (ignoring any financial considerations). At diagnosis, DNA methods can be used to both quickly determine breakpoints useful in designing DNA monitoring assays and other mutations that might influence initial response to TKI therapy. RNA sequencing (RNA seq) can used to assay specific genes and pathways associated with early response. Single-cell genetics can be used to establish potentially troublesome complex heterogeneity and identify populations with a resistance signature. During therapy, digital and DNA PCR can be used as a more sensitive determinant of deep molecular response, and “NGS+” used to identify mutations in patients without a deep response who appear to be relapsing.
Here are some potential uses of the new technology described.
Understanding the cancer ecosystem. Sensitive detection of mutations, coupled with single-cell genomics, will allow us to study how clonal architecture influences initial response to therapy and how it changes with therapy.53,54 The identification of what clones persist after therapy (at MRD) will allow for targeting of the resistant clones destined for relapse. Characterization of the immune system by single-cell methods can offer insights into why some patients with MRD relapse, whereas some do not. Currently, cancer uses natural selection against us, and these technologies may let us turn the tables, using natural selection against cancer.
Further defining MRD. As noted, not all MRD is created equal,55 and without more sensitive methods to detect lower amounts of MRD and to genetically characterize MRD, optimal ways to use MRD to guide therapy will remain elusive. New sequencing techniques are transferable to other cancers and could yield more accurate and sensitive MRD determinations compared with current methods.
Detection and characterization of clonal hematopoiesis of indeterminant prognosis (CHIP). Understanding CHIP (how soon it occurs, levels that are meaningful, what combinations of mutations are worrisome) will be greatly enhanced by more sensitive and comprehensive mutational analyses.56-58 Here, there is great potential using new sequencing methods and single-cell genotyping to better understand what CHIP means and how rapidly it evolves (moving from indeterminant to determinant).
Outreach to low-resource areas. The vast majority of the world’s cancer patients live in areas without access to cancer therapeutics and diagnostics. Innovations of inexpensive diagnostics in CML are advancing, and this should be the model for other diseases in which targeted agents can be wed with molecular diagnostic assays.
We are in a unique era in which the Venn diagram overlap of biotechnology, genetics, biology, and mathematics is constantly enlarging. If we overlay this with imagination and compassion, the field of cancer diagnostics will innovate and amaze. Let leukemia lead the way.
Authorship
Contribution: J.R. conceived the project; J.R., C.Y., and D.W. contributed to the writing; J.R. did final editing.
Conflict-of-interest disclosure: J.R. occasional consulting with Novartis and BMS advisory boards; twice-daily and proposal laboratory contracts with Novartis. The remaining authors declare no competing financial interests.
Correspondence: Jerald Radich, Fred Hutchinson Cancer Research Center, 1100 Fairview Ave North, D4-100, Seattle, WA 98109; e-mail: jradich@fredhutch.org.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal