

In this issue of Blood, provide new insights into the mechanisms responsible for the well-documented finding of thrombocytosis in patients with iron deficiency anemia. By using mouse and human models, they document that a low iron content in the bone marrow environment affects the megakaryocyte-erythroid progenitors (MEPs). This occurs because of induced alteration in their metabolism, attenuation of ERK signaling, slowing of cell proliferation, and biasing of the MEPs toward megakaryocyte commitment, which ultimately results in thrombocytosis and anemia.1
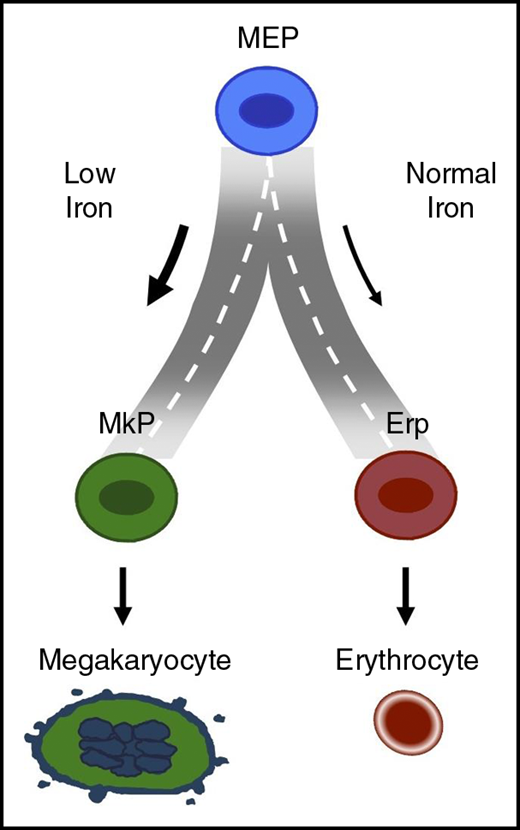
Model describing how iron may regulate the road taken by MEP when it arrives at the fork to choose between red cell vs platelet production.
Model describing how iron may regulate the road taken by MEP when it arrives at the fork to choose between red cell vs platelet production.
Iron deficiency is the most prevalent cause of anemia worldwide and is estimated to affect 1.5 billion people globally, primarily women and children.2 Iron deficiency anemia is associated with impaired neurocognitive development and immune dysfunction in younger children. Although nutritional deficiency has long been recognized as the primary cause of iron deficiency, recent studies have shed new insights into the regulation of iron homeostasis, which can also affect iron balance.3
Although anemia has long been recognized as the dominant clinical feature of iron deficiency, it has also been noted that patients often have elevated platelet counts.4 It is well known that inadequate access to iron for the synthesis of heme leads to a decrease in red cell hemoglobin content and red cell volume. In marked contrast, our understanding of the mechanistic basis for thrombocytosis in iron deficiency and the pathophysiological sequelae of this increase are less well defined.
In the generally accepted model of hematopoiesis, hierarchical differentiation of the self-renewing pluripotent hematopoietic stem cell (HSC) leads to the production of stem cells with limited capacity for self-renewal (short-term HSCs), which in turn give rise to multipotent progenitors that can commit to either the myeloid lineage or the lymphoid lineage. Ongoing work is shedding new light on the relative contributions of cytokines, growth factors, cell-cell interactions, transcriptional factor networks, epigenetics, and cell metabolism in determining cell fate decisions.5-7 Significant technical advances during the last decade, including the identification of a wide variety of new surface markers and the availability of high-speed flow sorters, have allowed researchers to isolate highly enriched populations of hematopoietic cells at various stages of development.8-10
On the basis of the surface expression of Lin–CD34+CD38midCD45RA–FLT3–MPL+CD36–CD41– in conjunction with flow cytometry-based cell sorting, a recent study described optimal strategies to obtain highly enriched populations of bipotent MEPs that can give rise to both megakaryocyte and erythroid lineages.10 During the course of that work, strategies were also developed for enriching populations of primary human lineage–committed megakaryocyte progenitors (MkPs) and erythroid progenitors (ErPs). These developments set the stage for the Xavier-Ferrucio study to critically assess how iron may act in decisions regarding fate of MEPs to follow the trajectory for making either megakaryocytes or erythroid cells.
By using 2 murine models of iron deficiency (dietary manipulation and mice with genetic defects that impair dietary iron uptake), the investigators provide convincing evidence that a low iron environment promotes megakaryocyte commitment of MEPs at the expense of erythroid commitment (see figure). The mechanistic basis for this iron-related switch in lineage commitment of MEPs was shown to be related to decreased cell proliferation and decreased ERK phosphorylation. The extensive data from a large series of murine models make a convincing case that iron regulates the lineage commitment choice of MEPs. Another important feature of the Xavier-Ferrucio study is the documentation that iron regulation of MEP commitment noted in murine studies also applies to human MEPs in vitro. The novel finding that iron levels can dictate lineage commitment of MEPs to either MkP or ErP implies that iron regulates the road taken by MEPs when they arrive at the fork to choose between red cell vs platelet production.
Although the reported findings represent an important step in our understanding of the role of iron in MEP lineage commitment, a number of questions remain unanswered. It is not clear why thrombocytosis occurs only in some patients with iron deficiency and not in others. It is possible that differences in the level of iron deficiency or in iron processing affect hematopoietic progenitor subpopulations. Given that genome-wide association studies have identified common genetic polymorphisms that influence hematologic traits such as hemoglobin levels and platelet counts, is it also possible that genetic factors have an impact on the pathway(s) mediating thrombocytosis in the setting of iron deficiency anemia. With regard to a potential evolutionary advantage, can iron deficiency, by limiting erythroid differentiation, preserve iron for other functions and/or that thrombocytosis could have conferred a selective advantage for individuals with iron deficiency anemia facing life-threatening hemorrhage? It is anticipated that answers to these important questions will be pursued in future studies.
In spite of some of these unanswered questions, the studies of Xavier-Ferrucio et al significantly advance our understanding of the regulation of lineage commitment by human hematopoietic progenitor cells. There are valuable insights about the role of not only growth factors and cytokines but also the regulation of cell metabolism and cell signaling by iron. It is likely that the experimental approaches described will be useful in furthering our understanding of the regulation of human hematopoiesis.
Conflict-of-interest disclosure: The authors declare no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal