


Abstract
DNA methylation has pivotal regulatory roles in mammalian development, retrotransposon silencing, genomic imprinting, X-chromosome inactivation, and cancer. Cancer cells display highly dysregulated DNA methylation profiles, characterized by global hypomethylation in conjunction with hypermethylation of promoter CpG islands; these changes are often correlated with promoter hypermethylation, leading to decreased expression of tumor suppressor genes, as well as with genome instability, leading to amplification and aberrant expression of oncogenes. Ten-eleven-translocation (TET) proteins are α-ketoglutarate (α-KG)–dependent dioxygenases that oxidize 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC) and the additional oxidation products 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC); together, these oxidized methylcytosines are intermediates in DNA demethylation. TET2 is frequently mutated in diverse lymphoid and myeloid cancers, and TET loss of function is often observed in the absence of coding region mutations in TET genes. Despite our understanding of the biochemical activities of TET proteins, how TET loss of function promotes the onset and progression of hematopoietic malignancies is largely unknown. Here, we review recent advances in our understanding of the role of TET enzymes in lymphoid and myeloid neoplasms and highlight the importance of metabolic alterations that decrease TET activity in cancer initiation and progression.
Introduction
Recent advances in genomic sequencing have identified numerous somatic gene mutations in cancers, many of which involve dysregulation of enzymes that introduce epigenetic modifications into chromatin. We focus here on mutations in the ten-eleven translocation (TET) family of epigenetic regulators, which alter the modification status of 5-methylcytosine (5mC) in DNA and promote DNA demethylation. We briefly describe recent findings regarding the function of TET proteins in lymphoid and myeloid cells; for additional details on the roles of TET enzymes in myeloid malignancies and immune cell function, we refer the reader to previous reviews.1-5 TET loss-of-function (LOF) can occur even in the absence of TET coding region mutations. In the last section of this review, we describe the emerging relationship between TET inhibition and cell transformation caused by a dysregulated metabolome.
DNA-modifying enzymes: DNMT and TET
5mC is generated by DNA methyltransferases (DNMTs), which transfer a methyl group from S-adenosylmethionine to the 5-position of cytosine bases6-8 (Figure 1). The vast majority of 5mC is located in CG (CpG) dinucleotides in DNA, which undergo symmetrical methylation on both DNA strands; however, certain CpG-rich regions, known as CpG islands (CGIs), are poorly methylated. During DNA replication, CpGs on newly synthesized DNA strands are unmethylated, but symmetrical methylation is rapidly restored by the maintenance methyltransferase DNMT1.6-8 DNMT1 acts in complex with its partner, UHRF1, which possesses an SRA domain that recognizes hemimethylated CpGs; symmetrical methylation is fully restored within 20 minutes of replication.9 In contrast, DNMT3A and DNMT3B catalyze the de novo methylation of unmethylated CpGs.6-8
![TET enzymes, DNA modification, and DNA demethylation. (A) TET-mediated 5mC oxidation. TET uses reduced iron (Fe2+), oxygen, and α-ketoglutarate (αKG) to oxidize 5mC, generating the products CO2, succinate, and the oxidized methylcytosine (oxi-mC) bases 5-hydroxymethyl (5hmC), 5-formyl (5fC), and 5-carboxylcytosine (5caC). The enzymatic activity of TET is modulated by the levels of αKG and is subject to product inhibition by succinate. Vitamin C enhances the enzymatic activity of TET, most likely by maintaining Fe2+ in its reduced state. Hypoxia and both enantiomers of the oncometabolite 2-hydroxyglutarate (2HG) inhibit TET activity. (B) TET2-DNA interaction.112 The diagram shows a portion of the TET2 catalytic domain (gray; amino acids [aa]1129-1480, 1844-1936) interacting with 5mC (orange) on double-stranded DNA (turquoise). The 5mC base is flipped and inserted into the TET2 active site with the methyl group adjacent to Fe2+ (blue) and αKG (magenta). Zinc ions are shown in yellow. (C) TET-mediated DNA demethylation. Unmodified cytosines in DNA are methylated by DNMTs to yield 5mC. TET proteins successively oxidize 5mC to 5hmC, 5fC, and 5caC. 5hmC is the most abundant of the oxi-mCs (∼1% to 10% of 5mC in most somatic cell types and often higher in neurons); 5fC and 5caC are ∼10- to 100-fold and ∼100- to 1000-fold less abundant, respectively, compared with 5hmC. Oxi-mCs present on the unreplicated DNA strand in the CpG sequence context are not recognized by the DNMT1/UHRF1 complex, which normally recognizes hemimethylated CpGs; this prevents the restoration of symmetrical methylation on the newly replicated strand and facilitates passive (replication-dependent) DNA demethylation (top arrow). TET can also facilitate DNA demethylation independently of DNA replication (bottom arrow), because 5fC and 5caC can both be excised by thymine DNA glycosylase (TDG) and replaced with unmodified cytosine through base-excision repair (BER). (D) TET2 mutations in DLBCL. Diagrammatic representation of the domain structure of TET2 including the cysteine-rich domain (cys-rich; aa1129-1312) and catalytic domain. Dotted line (aa1481-1843) represents the region replaced with low-complexity linker in the structural study in panel B (top).112 Hatch marks showing the distribution of TET2 mutations observed in diffuse large B-cell lymphoma (DLBCL; data from cBioPortal113,114). In a total of 1295 DLBCL cases, 72 (∼5.6%) were observed to bear TET2 mutations, of which 34 were nonsense mutations predicted to produce a truncated TET2 protein and 46 were missense mutations predicted to give rise to single amino acid substitutions (middle and bottom). Note that TET1 and TET3 mutations are rare (∼0.2%).](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/134/18/10.1182_blood.2019791475/2/m_bloodbld2019791475cf1.png?Expires=1770259182&Signature=qWh4y61AfdVEnceJzM7eQ4LXzV1bsO785u46rK7adhiCIiM82JL5Wtb3wWOM2D9HultIcmhzO06Y-A1obxnawAeUUM0JwKrQ-f1PJWv4Ty312g28gZQU9G9~JMMPhQaa45yd3pI2q1FqYq~tl-antnOZTRNGNcCBo62fmHapZhnPQvhB0TOJ8Bu9m~~S~gxkUvHqzpNgoDvpi930-TW5QIrfjd3DNqrqEOSd4J0pdlcMeYQx~BthH7ztfWKI~Fc42SBvvBJwbOGcjRfjMeMm~3qPSpbff~u5yjopD2IraiWjNsmj57iZinQ4z4yLy07-U0TWk5rBTvw8a8pWDmjUsA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
TET enzymes, DNA modification, and DNA demethylation. (A) TET-mediated 5mC oxidation. TET uses reduced iron (Fe2+), oxygen, and α-ketoglutarate (αKG) to oxidize 5mC, generating the products CO2, succinate, and the oxidized methylcytosine (oxi-mC) bases 5-hydroxymethyl (5hmC), 5-formyl (5fC), and 5-carboxylcytosine (5caC). The enzymatic activity of TET is modulated by the levels of αKG and is subject to product inhibition by succinate. Vitamin C enhances the enzymatic activity of TET, most likely by maintaining Fe2+ in its reduced state. Hypoxia and both enantiomers of the oncometabolite 2-hydroxyglutarate (2HG) inhibit TET activity. (B) TET2-DNA interaction.112 The diagram shows a portion of the TET2 catalytic domain (gray; amino acids [aa]1129-1480, 1844-1936) interacting with 5mC (orange) on double-stranded DNA (turquoise). The 5mC base is flipped and inserted into the TET2 active site with the methyl group adjacent to Fe2+ (blue) and αKG (magenta). Zinc ions are shown in yellow. (C) TET-mediated DNA demethylation. Unmodified cytosines in DNA are methylated by DNMTs to yield 5mC. TET proteins successively oxidize 5mC to 5hmC, 5fC, and 5caC. 5hmC is the most abundant of the oxi-mCs (∼1% to 10% of 5mC in most somatic cell types and often higher in neurons); 5fC and 5caC are ∼10- to 100-fold and ∼100- to 1000-fold less abundant, respectively, compared with 5hmC. Oxi-mCs present on the unreplicated DNA strand in the CpG sequence context are not recognized by the DNMT1/UHRF1 complex, which normally recognizes hemimethylated CpGs; this prevents the restoration of symmetrical methylation on the newly replicated strand and facilitates passive (replication-dependent) DNA demethylation (top arrow). TET can also facilitate DNA demethylation independently of DNA replication (bottom arrow), because 5fC and 5caC can both be excised by thymine DNA glycosylase (TDG) and replaced with unmodified cytosine through base-excision repair (BER). (D) TET2 mutations in DLBCL. Diagrammatic representation of the domain structure of TET2 including the cysteine-rich domain (cys-rich; aa1129-1312) and catalytic domain. Dotted line (aa1481-1843) represents the region replaced with low-complexity linker in the structural study in panel B (top).112 Hatch marks showing the distribution of TET2 mutations observed in diffuse large B-cell lymphoma (DLBCL; data from cBioPortal113,114 ). In a total of 1295 DLBCL cases, 72 (∼5.6%) were observed to bear TET2 mutations, of which 34 were nonsense mutations predicted to produce a truncated TET2 protein and 46 were missense mutations predicted to give rise to single amino acid substitutions (middle and bottom). Note that TET1 and TET3 mutations are rare (∼0.2%).
TET enzymes, DNA modification, and DNA demethylation. (A) TET-mediated 5mC oxidation. TET uses reduced iron (Fe2+), oxygen, and α-ketoglutarate (αKG) to oxidize 5mC, generating the products CO2, succinate, and the oxidized methylcytosine (oxi-mC) bases 5-hydroxymethyl (5hmC), 5-formyl (5fC), and 5-carboxylcytosine (5caC). The enzymatic activity of TET is modulated by the levels of αKG and is subject to product inhibition by succinate. Vitamin C enhances the enzymatic activity of TET, most likely by maintaining Fe2+ in its reduced state. Hypoxia and both enantiomers of the oncometabolite 2-hydroxyglutarate (2HG) inhibit TET activity. (B) TET2-DNA interaction.112 The diagram shows a portion of the TET2 catalytic domain (gray; amino acids [aa]1129-1480, 1844-1936) interacting with 5mC (orange) on double-stranded DNA (turquoise). The 5mC base is flipped and inserted into the TET2 active site with the methyl group adjacent to Fe2+ (blue) and αKG (magenta). Zinc ions are shown in yellow. (C) TET-mediated DNA demethylation. Unmodified cytosines in DNA are methylated by DNMTs to yield 5mC. TET proteins successively oxidize 5mC to 5hmC, 5fC, and 5caC. 5hmC is the most abundant of the oxi-mCs (∼1% to 10% of 5mC in most somatic cell types and often higher in neurons); 5fC and 5caC are ∼10- to 100-fold and ∼100- to 1000-fold less abundant, respectively, compared with 5hmC. Oxi-mCs present on the unreplicated DNA strand in the CpG sequence context are not recognized by the DNMT1/UHRF1 complex, which normally recognizes hemimethylated CpGs; this prevents the restoration of symmetrical methylation on the newly replicated strand and facilitates passive (replication-dependent) DNA demethylation (top arrow). TET can also facilitate DNA demethylation independently of DNA replication (bottom arrow), because 5fC and 5caC can both be excised by thymine DNA glycosylase (TDG) and replaced with unmodified cytosine through base-excision repair (BER). (D) TET2 mutations in DLBCL. Diagrammatic representation of the domain structure of TET2 including the cysteine-rich domain (cys-rich; aa1129-1312) and catalytic domain. Dotted line (aa1481-1843) represents the region replaced with low-complexity linker in the structural study in panel B (top).112 Hatch marks showing the distribution of TET2 mutations observed in diffuse large B-cell lymphoma (DLBCL; data from cBioPortal113,114 ). In a total of 1295 DLBCL cases, 72 (∼5.6%) were observed to bear TET2 mutations, of which 34 were nonsense mutations predicted to produce a truncated TET2 protein and 46 were missense mutations predicted to give rise to single amino acid substitutions (middle and bottom). Note that TET1 and TET3 mutations are rare (∼0.2%).
The mammalian TET family proteins TET1, TET2, and TET3 are dioxygenases that facilitate passive and active DNA demethylation. TET enzymes use molecular oxygen and the cofactors αKG (a metabolite of the tricarboxylic acid [TCA] cycle) and reduced Fe2+ to oxidize the methyl group of 5mC successively to 5hmC, 5fC, and 5caC10-13 (Figure 1A-C). All 3 oxidized methylcytosines (oxi-mCs) are intermediates in DNA demethylation (Figure 1C). Symmetrically methylated CpG motifs containing 5mC on both DNA strands become hemimethylated during DNA replication; however, if oxi-mC is present on the template strand, the unmethylated cytosine on the newly synthesized strand is not effectively recognized or methylated by the DNMT1/UHRF1 complex.14,15 This leads to the loss of DNA methylation as cells divide, in a process known as passive (replication-dependent) DNA demethylation (Figure 1C top). In addition, 5fC and 5caC can be removed from properly base-paired 5fC:G and 5caC:G base pairs by thymine DNA glycosylase, which normally excises T:G mismatches; the base-excision repair system then replaces the oxi-mCs with unmodified cytosines, a process known as active DNA demethylation (Figure 1C bottom). In most cell types investigated, passive DNA demethylation is the major mechanism by which TET proteins promote DNA demethylation.
TET2 mutations in human hematopoietic disorders
The TET2 gene is more frequently mutated in hematopoietic malignancies than genes encoding the other TET family members; the reason for this is not entirely clear, because TET1 and TET3 are also expressed in most hematopoietic lineages. There are no mutational hotspots in TET216 (Figure 1D); rather, nonsense and missense mutations are distributed across the TET2 coding region, indicating that mutations in TET2 are LOF mutations and that TET2 functions as a tumor suppressor.16
Clonal hematopoiesis
Clonal hematopoiesis of indeterminate prognosis (CHIP; also known as age-related clonal hematopoiesis [ARCH]) is defined as the aberrant expansion of clones of blood cells derived from a single hematopoietic stem cell (HSC; Figure 2). The predominant mutations associated with CHIP are DNMT3A, TET2, and ASXL1.17-19 The incidence of CHIP increases with age, but many individuals with CHIP are clinically normal; nevertheless, clonal hematopoiesis is likely to be a premalignant state that can be identified many years before the development of a clinically apparent disorder, such as myelodysplastic syndrome (MDS) or secondary AML.19
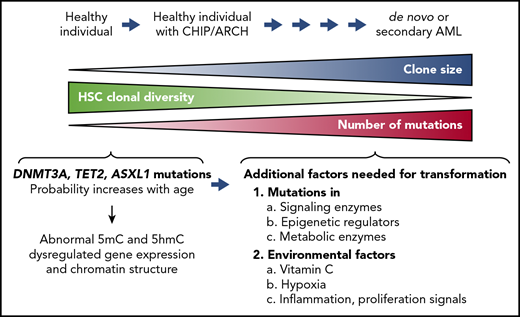
Clonal hematopoiesis. The diagram illustrates disease progression from clonal hematopoiesis to acute myeloid leukemia (AML). Somatic LOF mutations of TET2, DNMT3A, or ASXL1 confer a competitive advantage on HSCs and result in aberrant expansion of mutant HSC clones. This process, also known as CHIP or ARCH, can be observed in healthy individuals who may not develop clinical abnormalities for decades. The clonal diversity of HSCs contributing to hematopoiesis gradually decreases in individuals with CHIP/ARCH, concomitantly with an increase in the size of the HSC clones bearing the mutations. Secondary mutations tend to accumulate in these mutated HSCs with age, predisposing individuals with CHIP/ARCH to a higher risk of developing hematopoietic malignancies, including de novo or secondary AML. Additional factors that could contribute to cell transformation are mutations in signaling enzymes and/or epigenetic regulators, metabolic alterations, and environmental factors, including vitamin C levels, inflammation, and hypoxia (lower right).
Clonal hematopoiesis. The diagram illustrates disease progression from clonal hematopoiesis to acute myeloid leukemia (AML). Somatic LOF mutations of TET2, DNMT3A, or ASXL1 confer a competitive advantage on HSCs and result in aberrant expansion of mutant HSC clones. This process, also known as CHIP or ARCH, can be observed in healthy individuals who may not develop clinical abnormalities for decades. The clonal diversity of HSCs contributing to hematopoiesis gradually decreases in individuals with CHIP/ARCH, concomitantly with an increase in the size of the HSC clones bearing the mutations. Secondary mutations tend to accumulate in these mutated HSCs with age, predisposing individuals with CHIP/ARCH to a higher risk of developing hematopoietic malignancies, including de novo or secondary AML. Additional factors that could contribute to cell transformation are mutations in signaling enzymes and/or epigenetic regulators, metabolic alterations, and environmental factors, including vitamin C levels, inflammation, and hypoxia (lower right).
Large-scale prospective studies involving >100 000 healthy persons were recently performed to examine the occurrence of somatic mutations before the development of AML.20-22 Mutually exclusive, LOF somatic mutations in DNMT3A (19%) and TET2 (6%) were observed a long time before AML onset in healthy persons with CHIP; in contrast, a significant percentage of AML patients had both DNMT3A and TET2 mutations,20 emphasizing the major contribution of the DNA modification pathway to leukemogenesis. Thus, DNMT3A or TET2 mutation alone seems sufficient to induce CHIP/ARCH in healthy individuals, but mutations in both genes may greatly facilitate AML. These observations are consistent with a recent study of mice doubly deficient for both Dnmt3a and Tet2.23
Myeloid malignancies
Somatic mutations of the TET2 gene have been observed in various myeloid disorders, including myeloproliferative neoplasms (MPNs; ∼13%), MDS (25%), chronic myelomonocytic leukemia (CMML; ∼50%), and AML (∼23%).2,18,24-28 Patients with TET2 mutations have a longer interval before the clinical onset of leukemia compared with patients with other mutations, such as internal tandem duplication (ITD) mutations in FLT3 (FLT3ITD)20,21,29 ; moreover, the TET2 mutational burden is higher in chronic compared with acute disease,2,24-26,28-30 suggesting that TET2 mutations cannot drive efficient leukemogenesis alone but instead require additional mutations in genes such as KRAS, RUNX1, ASXL1, ZRSR2, KIT, SMC3, CBL, EZH2, and CUL.11,12,29 In myeloid leukemias, mutations in the IDH1 or IDH2 genes are largely mutually exclusive with TET2 mutations,27,31,32 but this is not true of lymphoid malignancies.
T-cell malignancies
Peripheral T-cell lymphomas (PTCLs) are a group of aggressive lymphomas mostly derived from T cells that account for 12% to 15% of all non-Hodgkin lymphomas. Two of the most common subtypes of PTCL include PTCL not otherwise specified (PTCL-NOS; 26%) and angioimmunoblastic T-cell lymphoma (AITL; ∼20%).33,34 On the basis of gene expression signatures, most AITLs and ∼20% of PTCL-NOSs are likely derived from follicular helper T (Tfh) cells, the T cells that facilitate B-cell antibody responses by interacting with B cells in the germinal center.35,36 The 5-year overall survival rate is only 30% for AITL,34 demonstrating the aggressiveness of this lymphoma.
TET2 mutations are frequently found in T-cell malignancies.37-40 In 1 study, 10 (33.3%) of 30 AITL patients and 6 (20%) of 30 PTCL-NOS patients harbored TET2 mutations.37 The aggregated TET2 mutation frequencies in AITL and PTCL-NOS from a total of 3 studies were 63% (127 of 201) and 36% (27 of 75), respectively.38-40 TET2 mutation recurrently co-occurs with DNMT3A, IDH2, and RHOA G17V mutations; in fact, RHOA was mutated in ∼68% of AITL patients, and TET2 was comutated in all cases,34,41 again suggesting that the TET2 mutation represents the initial mutagenic event and predisposes the cells to lymphomagenesis caused by subsequent mutations such as RHOA.
B-cell malignancies
TET2 mutations are also frequently found in B-cell lymphomas, particularly in DLBCL. DLBCL is the most frequent type of non-Hodgkin lymphoma originating from germinal-center B cells. Mutational analyses of DLBCL showed that TET2 was mutated in ∼6% to 12% of samples,37,42 a result confirmed by recent large-scale analyses of >1000 DLBCL patients.43,44 TET1 coding region mutations are rarely found in B-cell lymphomas, but TET1 was shown to be epigenetically silenced in several cases of follicular lymphoma, DLBCL, and multiple myeloma (MM).45 A study of 574 samples by Staudt et al44 showed that DLBCL could be categorized into 4 groups based on mutation profiles; of samples categorized as unclassified, ∼10% had TET2 mutations. This category of DLBCL may be enriched for TET LOF, whether or not TET2 coding mutations are observed.
Mouse experimental models to explore the role of TET LOF in oncogenesis
In human cancers, genetic heterogeneity and diverse and complicated gene alteration patterns, specifically the multiple polymorphisms and somatic mutations observed in any given malignancy, make it difficult to determine the impact of any individual mutation or combination of mutations. The advantages of mouse models are the inbred genetic background and the relative simplicity of mutational patterns imposed by the researcher; mutations can be designed to mimic recurrent alterations (whether LOF or gain-of-function [GOF]) or capture combinations of mutations that occur frequently in human malignancies. Thus, mouse models are a first step in understanding the underlying mechanisms of oncogenesis in human cancers, particularly how different mutations cooperate with one another. A summary of the phenotypes of Tet-deficient mouse models with known median survival is shown in Figure 3 and Table 1.
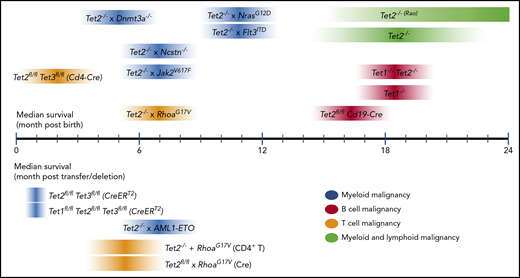
Median survival times reported for mouse models of Tet deficiency. Median survival reported for models with germ line– or cell type–specific transgenes or gene deletions (top); median survival reported for inducible gene disruption or adoptive cell transfer models (bottom). For references, please refer to Table 1. For each Tet-deficient mouse model, the darkest color at the midpoint of each ribbon represents the approximate median survival. Note that the width and gradation of color do not necessarily reflect any statistical property.
Median survival times reported for mouse models of Tet deficiency. Median survival reported for models with germ line– or cell type–specific transgenes or gene deletions (top); median survival reported for inducible gene disruption or adoptive cell transfer models (bottom). For references, please refer to Table 1. For each Tet-deficient mouse model, the darkest color at the midpoint of each ribbon represents the approximate median survival. Note that the width and gradation of color do not necessarily reflect any statistical property.
Median survival of Tet-deficient animal models
| Type of malignancy | Median survival, mo | Reference |
|---|---|---|
| Myeloid and lymphoid | ||
| Tet2−/− | 12-18 | 46,51,59 |
| Tet2−/− | >24 | Unpublished (A.R.) |
| Myeloid | ||
| Tet2fl/flTet3fl/fl (CreERT2; acute deletion) | 1 | 115 |
| Tet1fl/flTet2fl/flTet3fl/fl (CreERT2; acute deletion) | 1 | Unpublished (A.R.) |
| Tet2−/−Dnmt3a−/− | 5 | 23 |
| Tet2−/−Jak2V617F | 6 | 116 |
| Tet2−/−AML1-ETO | 6 | 117 |
| Tet2−/−Ncstn−/− | 6 | 118 |
| Tet2−/−NrasG12D | 9-12 | 119 |
| Tet2−/−FLT3ITD | 9-12 | 27 |
| T cell | ||
| Tet2fl/flTet3fl/fl (CD4-Cre) | 2 | 50 |
| Tet2−/−RhoaG17V (T-cell transfer) | 5 | 53 |
| Tet2fl/flRhoaG17V (Cd4-CreERT2 inducible) | 6 | 54 |
| Tet2fl/fl (Vav-Cre) Cd4-RhoaG17V (transgene) | 7 | 55 |
| B cell | ||
| Tet2fl/flCd19-Cre | 16 | 60 |
| Tet1−/− | 22 | 45 |
| Tet1−/−Tet2−/− | 20 | 59 |
| Type of malignancy | Median survival, mo | Reference |
|---|---|---|
| Myeloid and lymphoid | ||
| Tet2−/− | 12-18 | 46,51,59 |
| Tet2−/− | >24 | Unpublished (A.R.) |
| Myeloid | ||
| Tet2fl/flTet3fl/fl (CreERT2; acute deletion) | 1 | 115 |
| Tet1fl/flTet2fl/flTet3fl/fl (CreERT2; acute deletion) | 1 | Unpublished (A.R.) |
| Tet2−/−Dnmt3a−/− | 5 | 23 |
| Tet2−/−Jak2V617F | 6 | 116 |
| Tet2−/−AML1-ETO | 6 | 117 |
| Tet2−/−Ncstn−/− | 6 | 118 |
| Tet2−/−NrasG12D | 9-12 | 119 |
| Tet2−/−FLT3ITD | 9-12 | 27 |
| T cell | ||
| Tet2fl/flTet3fl/fl (CD4-Cre) | 2 | 50 |
| Tet2−/−RhoaG17V (T-cell transfer) | 5 | 53 |
| Tet2fl/flRhoaG17V (Cd4-CreERT2 inducible) | 6 | 54 |
| Tet2fl/fl (Vav-Cre) Cd4-RhoaG17V (transgene) | 7 | 55 |
| B cell | ||
| Tet2fl/flCd19-Cre | 16 | 60 |
| Tet1−/− | 22 | 45 |
| Tet1−/−Tet2−/− | 20 | 59 |
Myeloid malignancies
Tet2-deficient mice develop both myeloid and lymphoid malignancies, usually after long latencies (∼2 years), and the myeloid leukemias accumulate mutations in many genes recurrently deleted or mutated in human hematological malignancies (eg, Apc, Nf1, Flt3, Cbl, Notch, Mll2).46 Tet2f/fVav1-Cre mice, which show Tet2 inactivation in all hematopoietic cells, including HSCs, had increased white blood cell counts and dramatically increased proportions of Gr1+/Mac1+ cells in their bone marrow (BM), spleen, and peripheral blood by 12 to 14 months; this was not observed in Tet2f/fLysM-Cre mice,47 in which Tet2 was inactivated in differentiated myeloid lineage cells. Thus, deletion of Tet2 in differentiated myeloid cells is not sufficient to cause myeloid malignancy in mice; rather, Tet2 deletion needs to occur in early precursor HSCs.
Inflammatory signals may enhance malignancy in TET-deficient mice, which show upregulation of several inflammatory mediators, including interleukin-6.48 Aged Tet2−/− mice displayed a preleukemic myeloproliferative syndrome induced as a result of increased bacterial translocation across gut mucosa and increased interleukin-6 production.49 This syndrome was reversed by antibiotic treatment and failed to develop in germ-free Tet2−/− mice, highlighting the importance of microbial signals in tumor development and potentially explaining the variable phenotypes observed in different strains of Tet2-deficient mice.2 Together, these data point to a role for TET2 in resolving inflammation in mice and suggest that increased production of inflammatory mediators could exacerbate leukemia development in TET-deficient settings.
T-cell malignancies
To analyze the in vivo function of TET proteins in T cells, our laboratory used Cd4-Cre to delete 2 functionally redundant TET proteins, Tet2 and Tet3, during T-cell thymic development.50 Tet2/3 double-deficient mice developed an aggressive PTCL-like syndrome that was apparent by 5 to 6 weeks, with all mice dying at ∼8 weeks of age; the malignant cells originated from invariant natural killer (NK) T cells in the thymus rather than from Tfh cells. Expansion required mitogenic signals delivered by the T-cell receptor, consistent with the signal-dependent myeloproliferation described for Tet2−/− mice.49 A different mouse strain (Tet2gt), in which Tet2 was targeted by a gene trap inserted at the second intron, developed lymphoproliferation of Tfh-like cells with a long latency (∼17 months).51 In humans, extranodal NK/T-cell lymphoma and some PTCL-NOS also express NK and NK T-cell markers, whereas Tfh markers are more frequent in AITL.35
As mentioned, TET2 and RHOA G17V mutations often co-occur in human AITL; RHOA is a small GTPase, and the G17V mutation encodes a dominant-negative protein that sequesters guanine-nucleotide exchange factors and is unable to bind GTP.40,41,52 Retroviral transduction of Tet2-deficient T cells with RHOAG17V led to massive proliferation of T cells bearing both mutations after transfer into T-cell lymphopenic Tcra−/− hosts; mice that received Tet2−/−RhoAG17V T cells died as a result of inflammatory diseases or developed aggressive cancer, with a median survival of 21.3 weeks.53 The mutations affected CD4 but not CD8 T cells and were associated with enhanced cell survival, increased Tfh numbers, and decreased regulatory T-cell differentiation. Cells with combined Tet2 deficiency and mutant RHOAG17V showed a cooperative decrease of FOXO1 expression and activity. FOXO1 reexpression reversed the defects in Tet2−/−RhoAG17V T cells, suggesting that FOXO1 is one of the prime targets of TET2 and RHOA in mouse CD4 T cells. In a second mouse strain, in which RHOAG17V was inducibly expressed from the endogenous Rhoa locus using Cd4-CreERT2, CD4 T cells preferentially differentiated into Tfh lineage cells, and after tamoxifen treatment followed by repeated immunization, mice receiving transplants of BM from RHOAG17VCd4-CreERT2 on Tet2−/− background developed similarly aggressive AITL-like lymphomas (median survival, 25 weeks).54 In a third model, transgenic mice expressing RHOAG17V under the control of CD4 regulatory elements55 showed impaired T-cell maturation and peripheral T-cell lymphopenia and developed autoimmune syndromes at ∼8 to 10 weeks, with Tfh cell expansion and autoantibody generation. RHOAG17V transgenic mice bearing an OT-II T-cell receptor transgene and with conditional deletion of Tet2 in the hematopoietic system using Vav1-Cre developed AITL-like lymphomas, with a median survival of 205 days, compared with 314 days for non-RHOAG17V controls.55 These studies suggest that TET2 and RHOA cooperatively regulate Tfh differentiation and demonstrate a synergistic interaction between Tet2 deficiency and other mutations such as RHOAG17V in T-cell lymphoma development.
In addition to being a tumor suppressor, TET2 also regulates peripheral CD8 T-cell differentiation. Consistent with the notion that TET2 and TET3 function redundantly,45,50,56 deletion of Tet2 alone using Cd4-Cre had minimal effect on thymic T-cell development. However, when Cd4-Cre Tet2-deficient mice were infected with lymphocytic choriomeningitis virus, the number of virus-specific CD8 T cells with memory phenotype increased, and these cells conferred improved protection upon subsequent infection.57 Whether Tet2 deficiency resulted in T-cell malignancy in this model is unclear, but the results reinforce the notion that TET2 limits cell proliferation and/or survival after mitogenic stimulation.
B-cell malignancies
TET proteins are essential for B-cell development and tumor suppression. When Mb1-Cre was used to delete Tet2 and Tet3 in early B cells, the cells displayed a developmental block at the transition from the pro–B-cell to the pre–B-cell stage56,58 but nonetheless developed a B-cell malignancy by 5 to 6 months that resembled B-cell acute lymphocytic leukemia (B-ALL).56 Similarly, mice deficient in both Tet1 and Tet2 developed leukemia resembling B-ALL, with long latency (median survival, 20 months), an observation consistent with the frequent downregulation of TET1 and TET2 expression in B-ALL patients.59 Tet1 deficiency conferred increased self-renewal capacity on mouse B-cell progenitors in vitro and resulted in B-cell lymphomas in vivo, with long latency (22 months).45
Among B-cell malignancies, TET2 mutations are most frequently found in DLBCL. Tet2 deficiency, induced using Cd19-Cre in B cells, resulted in expansion of B1a cells, and the mice eventually developed a B-cell malignancy similar to chronic lymphocytic leukemia.60 Exome sequencing revealed a pattern of activation-induced deaminase (AID)–mediated cytosine deamination, and leukemogenesis in these mice required AID.60 Intriguingly, maximal AID messenger RNA expression required TET-mediated DNA modification at the Aicda superenhancer,61 suggesting a potential failsafe mechanism to minimize AID-induced mutations in the absence of TET. Moreover, Tet2 mutations also precipitated malignancy induced by T-cell leukemia/lymphoma 1A (TCL1A), an oncogene relevant in both B and T cells.60 Taken together, these studies indicate that TET2 mutations create a permissive environment for mutations and lymphomagenesis.
Consistent with recurring TET2 mutations in DLBCL, TET mutations in mouse peripheral B cells resulted in germinal center (GC) B-cell hyperplasia and impaired plasma cell differentiation (C.-W.J.L. and A.R., unpublished data).62 Loss of Tet2 selectively affected 5hmC modification at the enhancers of genes related to GC exit and decreased the expression of Prdm1, the essential transcription factor for plasma-cell differentiation.62 Cells isolated from DLBCL patients with TET2 mutations exhibited gene expression signatures similar to those with CREBBP mutations, and TET2 and CREBBP mutations were mutually exclusive in DLBCL, suggesting that TET2 and CREBBP cooperate to regulate cell differentiation and cell-cycle exit and to prevent lymphomagenesis in GC B cells.62
The relative paucity of TET2 mutations in lymphoid compared with myeloid malignancies may reflect the existence of other mechanisms affecting TET activity. For instance, although TET coding mutations are rarely found in MM, an enhancer hypermethylation phenotype has been observed in MM,63 consistent with the decrease of TET1 expression observed in MM.45
TET LOF without TET coding mutations: metabolic alterations that decrease TET activity
Even in the absence of TET coding region mutations, tumors can exhibit impaired TET expression or function. When 5hmC, a surrogate for TET enzymatic activity, was measured in patients with diverse hematologic malignancies (MDS/MPN, CMML, AML), samples with TET2 mutations invariably had low 5hmC as expected, whereas samples with wild-type TET2 (and TET1 and TET3) showed a spread of 5hmC ranging from normal to low.11 Many solid tumors, including melanoma and breast, liver, lung, and colon cancers, also have low 5hmC,2,16 a feature that could reflect 5hmC dilution as a result of rapid proliferation,64 but this may also be due to decreased expression or stability of TET messenger RNA or protein or decreased enzymatic activity.2,16
Alterations in metabolic enzymes and pathways are a major cause of decreased TET activity. The enzymatic activity of TET enzymes and other αKG-dependent dioxygenases requires molecular oxygen, reduced Fe2+, and αKG; in consequence, TET activity is sensitive to hypoxia (which in consequence is associated with DNA hypermethylation at the promoters of tumor suppresser genes65 ) and to mutations or altered expression of various metabolic enzymes that decrease αKG levels and/or increase the levels of 2HG, a so-called oncometabolite that inhibits dioxygenase activity by competing with αKG66-69 (Figure 4). There are 2 enantiomers of 2HG, d-2hg (also known as [r]-2hg) and L-2HG (also known as [s]-2hg), both normal metabolites present at relatively low levels in healthy cells. L-2HG is considerably more potent than d-2HG at inhibiting αKG-dependent dioxygenases,70,71 but overproduction of either enantiomer in cancer, as well as increased production of L-2HG in hypoxia and activated CD8+ T cells,72-74 can profoundly inhibit the activity of αKG-dependent dioxygenases.67,69
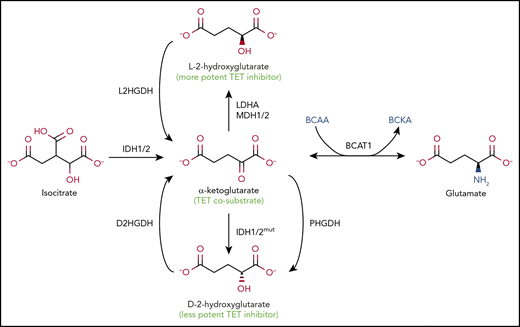
Regulation of TET enzymatic activity by metabolites. Metabolic pathways that alter levels of αKG and 2HG. The TET cosubstrate αKG, a TCA cycle intermediate, is produced from isocitrate by the cytoplasmic and mitochondrial isocitrate dehydrogenases IDH1 and IDH2, respectively (left). 2HG, a metabolite structurally similar to αKG, inhibits TET activity (middle). 2HG has 2 stereoisomers, L-2HG, also known as S-2HG (top), and D-2HG, also known as R-2HG (bottom), which are converted back to αKG by L-2-hydroxyglutarate dehydrogenase (L2HGDH) and D-2-hydroxyglutarate dehydrogenase (D2HGDH), respectively. GOF IDH mutations found in glioma, AML, and AITL can convert αKG into D-2HG, the prototype oncometabolite, a less potent inhibitor of TETs and other dioxygenases compared with L-2HG. Endogenous enzymes, such as phosphoglycerate dehydrogenase (PHGDH), an enzyme frequently amplified in breast cancers and melanoma, can also produce D-2HG, although it is not clear whether physiologically relevant concentrations of the metabolite are achieved. The more potent TET inhibitor L-2HG is generated by endogenous enzymes lactate dehydrogenase A (LDHA) and malate dehydrogenases 1 and 2 (MDH1/2). The level of αKG critically affects the activity of TET and other dioxygenases. The enzyme branched-chain amino acid (BCAA) transaminase 1 (BCAT1) reversibly transfers the amino group from the BCAAs leucine, isoleucine, and valine to αKG to yield branched-chain α-keto acids (BCKAs) and glutamate (right). The high levels of BCAT1 found in numerous cancers result in decreased levels of αKG, thus interfering with αKG-dependent enzymes, including TET proteins.
Regulation of TET enzymatic activity by metabolites. Metabolic pathways that alter levels of αKG and 2HG. The TET cosubstrate αKG, a TCA cycle intermediate, is produced from isocitrate by the cytoplasmic and mitochondrial isocitrate dehydrogenases IDH1 and IDH2, respectively (left). 2HG, a metabolite structurally similar to αKG, inhibits TET activity (middle). 2HG has 2 stereoisomers, L-2HG, also known as S-2HG (top), and D-2HG, also known as R-2HG (bottom), which are converted back to αKG by L-2-hydroxyglutarate dehydrogenase (L2HGDH) and D-2-hydroxyglutarate dehydrogenase (D2HGDH), respectively. GOF IDH mutations found in glioma, AML, and AITL can convert αKG into D-2HG, the prototype oncometabolite, a less potent inhibitor of TETs and other dioxygenases compared with L-2HG. Endogenous enzymes, such as phosphoglycerate dehydrogenase (PHGDH), an enzyme frequently amplified in breast cancers and melanoma, can also produce D-2HG, although it is not clear whether physiologically relevant concentrations of the metabolite are achieved. The more potent TET inhibitor L-2HG is generated by endogenous enzymes lactate dehydrogenase A (LDHA) and malate dehydrogenases 1 and 2 (MDH1/2). The level of αKG critically affects the activity of TET and other dioxygenases. The enzyme branched-chain amino acid (BCAA) transaminase 1 (BCAT1) reversibly transfers the amino group from the BCAAs leucine, isoleucine, and valine to αKG to yield branched-chain α-keto acids (BCKAs) and glutamate (right). The high levels of BCAT1 found in numerous cancers result in decreased levels of αKG, thus interfering with αKG-dependent enzymes, including TET proteins.
Mutant IDH enzymes
The 3 isocitrate dehydrogenases IDH1, IDH2, and IDH3 are encoded by 5 genes (IDH1, IDH2, IDH3A, IDH3B, and IDH3G) and catalyze the oxidative decarboxylation of isocitrate into αKG (Figure 4). IDH1 is cytoplasmic, whereas IDH2 and IDH3 function in the mitochondrial TCA pathway. Dominant mutations in key arginine residues in the active sites of IDH1 and IDH2 (R132 of IDH1, and R172 and R140 of IDH2) are recurrently observed in gliomas and AML.66-69 The mutant IDH enzymes use αKG, produced by IDH encoded on wild-type alleles in the same cell, to massively overproduce d-2HG, achieving intracellular concentrations of up to 10s of millimolars; these high D-2HG concentrations are likely needed for effective competition with αKG.66-69,75-77
Although 2HG inhibits the activity of all αKG-dependent dioxygenases, there is a clear correspondence between TET2 LOF and IDH GOF mutations: IDH1/2 and TET2 mutations both tend to be early events in leukemogenesis25,78 ; TET2- and IDH-mutant hematopoietic cells show similar DNA hypermethylation signatures and are associated with decreased 5hmC32,79 ; in MDS and AML, IDH1/2 GOF and TET2 LOF mutations coexist with similar secondary mutations, for instance, in FLT3, NPM1, and RAS25,32,80,81 ; and Tet2 knockout mice and Idh1R132 knock-in mice show parallel phenotypes, among them dysregulated DNA methylation, genome-wide decrease in 5hmC, impaired differentiation of human stem/precursor cells, myeloid skewing, and progression to myeloid malignancies.82,83 There are also substantial differences: TET2 mutations are more common in MDS/MPNs and CMML compared with AML, whereas IDH1/2 mutations are more frequent in de novo AML than in MDS/MPNs28,30 or in chronic myeloid neoplasms84 ; in MDS, IDH1 mutations are associated with shorter leukemia-free survival and decreased overall survival, whereas these features are not found in TET2-mutated MDS85,86 ; and unlike DNMT3A and TET2 mutations, IDH1/2 mutations are quite rare in clonal hematopoiesis (CHIP/ARCH).17,19 Overall, compared with TET2 mutations, IDH1/2 mutations tend to correlate with more aggressive disease.28,30 This might be explained by the fact that D-2HG overproduced by mutant IDH inhibits not only TET2 but also other αKG-dependent enzymes, including TET1 and TET3, JmjC-domain histone demethylases, and prolyl hydroxylases in the hypoxia pathway.
IDH1/2 and TET2 mutations tend to be mutually exclusive in myeloid neoplasms32 but not in AITL, one of the most common T-cell lymphomas. AITLs often exhibit TET2 (50%) or IDH2 mutations (20% to 30%), and TET2 mutations are present in 60% to 100% of IDH2R172-mutated AITLs.41,87 Therefore, even if TET2 and IDH1/2 mutations share a common oncogenic mechanism involving DNA modification, they also have at least some differing tumorigenic effects. Potentially, this could reflect a TET2-independent effect of IDH mutations on DNA repair through downregulation of ATM.31,88
Inhibitors selective for mutant IDH2 (enasidenib) and mutant IDH1 (ivosidenib) were approved by the US Food and Drug Administration in 201789,90 and 2018,91 respectively, for adult patients with relapsed or refractory AML with IDH1/2 genetic mutations. After a median follow-up of 8.3 months, 19.3% of enasidenib-treated patients experienced complete remission (CR), with an overall survival of 19.7 months,89 and 32.8% of ivosidenib-treated patients experienced CR or CR with partial hematologic recovery.91 Before therapy, IDH2-mutant clones showed variable differentiation arrest, but enasidenib treatment promoted hematopoietic differentiation from either terminal or ancestral mutant clones.92 The drugs prolong patient lifespan, but there are reports of acquired resistance to enasidenib by additional Q316E or I319M IDH2 mutations.93
2HG dehydrogenases
The enzymes L-2-hydroxyglutarate dehydrogenase (L2HGDH) and D-2-hydroxyglutarate dehydrogenase (D2HGDH) limit 2HG levels in cells by converting the corresponding enantiomers to αKG94 (Figure 4). Although germ line L2HGDH LOF mutation results in L-2HG acidurias with neuronal abnormality, somatic LOF mutations of L2HGDH are recurrent in renal cell carcinomas,95 and inactivating mutations of D2HGDH have been reported in a small subset (∼5%) of DLBCL patients.96 Experiments in cell lines suggested that the major cellular consequence of D2HGDH mutations was not increased D-2HG but rather decreased expression of mitochondrial IDH2, leading to decreased αKG.96
Cancer-associated mutations of other metabolic enzymes that increase 2HG or decrease αKG
At least 2 metabolic enzymes, LDHA and MDH1/2, facilitate the conversion of αKG to L-2HG,73,97 whereas phosphoglycerate dehydrogenase can catalyze the conversion of αKG to D-2HG98 (Figure 4). Increased expression of the hypoxia-inducible factor (HIF) target genes LDHA and MDH1/2 is a characteristic feature of hypoxia, and hypoxia can compromise TET activity by facilitating L-2HG generation via LDHA and MDH1/2.73,74 Altered expression of these enzymes may contribute to oncogenesis through inhibition of TET and other αKG-dependent dioxygenases.
The levels of αKG in cells can critically affect TET activity. Overexpression of the BCAT1 has been implicated in several aggressive tumors, including AML.99 In the cytosol, BCAT1 reversibly transfers the α-amino group from the BCAAs leucine, valine, and isoleucine to αKG, generating the corresponding branched-chain α-keto acid and glutamate (Figure 4). Proteomic analysis of primary AML stem cells compared with nonstem cells from human patients revealed high expression of BCAT1, and short hairpinRNA–mediated knockdown of BCAT1 decreased the growth and engraftment of these cells both in vitro and in vivo.99 Metabolic analyses of AML cell lines indicated that a major effect of high BCAT1 expression was to decrease αKG levels and thereby the activities of αKG-dependent dioxygenases, including TET and EGLN1/PHD2 (prolyl hydroxylases that regulate expression of HIF1α). Decreased TET activity correlated with decreased 5hmC and was associated with a DNA hypermethylation phenotype similar to that observed in IDH1/2-mutant AML.99
A similar increase in BCAT1 was observed in blast crisis compared with chronic-phase CML as well as in de novo AML100 ; in this case, however, the authors suggested that the phenotype reflected increased BCAA levels and increased activity of the mammalian target of rapamycin, presumably via leucine sensing. They also implicated the RNA binding protein Musashi2, which is massively upregulated in blast crisis over chronic-phase CML,101 in binding to and stabilizing the BCAT1 transcript, resulting in increased BCAT1 protein expression.100 Both mechanisms may apply, because high BCAT1 expression may drive the transamination reaction in either direction depending on intracellular reactant concentrations and cell context (Figure 4).
TET activity is intimately linked to the metabolites in the TCA cycle, and mutations of TCA-cycle enzymes have been linked to cancers. For instance, LOF mutations of fumarate hydratase or succinate dehydrogenase result in the accumulation of fumarate and succinate, respectively. These metabolites inhibit the activity of many αKG-dependent enzymes, including TET enzymes, JmjC lysine demethylases, and HIF prolyl hydroxylases, potentially through competitive inhibition of αKG by fumarate and product inhibition by succinate.102,103 In summary, dysregulated metabolic pathways could contribute directly to cancer progression by inhibiting αKG-dependent dioxygenases, including TET enzymes. However, the extent to which these enzymes are dysregulated in hematopoietic malignancies is currently unclear.
The TET activator vitamin C
As a cofactor for numerous αKG-dependent dioxygenases that most likely acts by maintaining Fe2+ in its reduced state, vitamin C (ascorbate) modulates the function of TET proteins and JmjC histone lysine demethylases.104 Vitamin C promotes the activity of TET enzymes in embryonic stem cells105 and in regulatory T cells,106,107 where it acts through TET2/3 to increase the stability of FOXP3 expression. Vitamin C also influenced the effect of mutant IDH1R132H and induced the expression of genes involved in leukocyte differentiation.108
Vitamin C is transported into cells by the transporter SLC23A2 and is enriched in both mouse and human HSCs and multipotent progenitors.109 Although humans are unable to synthesize vitamin C, mice can generate vitamin C through the action of the enzyme gulonolactone oxidase (GULO). Like humans, Gulo-deficient mice require dietary vitamin C for viability, but newborn Gulo−/− mice can survive if fostered by heterozygous mothers. Gulo−/− mice developed increased numbers of HSCs that displayed decreased 5hmC levels, and these features were similar to those observed in Tet2−/− as well as Tet2−/−Gulo−/− mice, indicating that vitamin C acted primarily through TET2 to cause HSC expansion.
In humans, TET2 and FLT3ITD mutations cooperate to cause AML, and this phenomenon has been replicated in mouse models. The role of vitamin C in the development of AML was examined using BM cells from FLT3ITD mice. When BM cells from Flt3ITD and wild-type mice were cotransferred into Gulo−/− recipients, the vitamin C–depleted environment promoted preferential myelopoiesis of Flt3ITD donor HSCs. The effect of vitamin C on myelopoiesis was dependent on TET2, because it was not observed when Tet2−/−FLT3ITD BM cells were transplanted; however, vitamin C potentially has additional tumor-suppressive function, because the Gulo−/− recipients developed leukemia with an accelerated onset. Lastly, depending on whether supplementation occurred before or after leukemia development, vitamin C could either completely or partially rescue the survival of Gulo−/− recipients transferred with BM cells from TET2-haploinsufficient (Tet2+/−) Flt3ITDmice.109
The aberrant self-renewal of Tet2-mutant HSCs was blocked by treatment with vitamin C, mimicking the effect of reconstitution with wild-type TET2.110 Specifically, vitamin C reduced the colony-formation capacity of Tet2-mutant BM cells but did not affect colony formation in double Tet2/3-deleted BM cells, indicating that vitamin C acts largely through these 2 TET proteins in this system. In a panel of AML cell lines, vitamin C induced DNA demethylation and gene expression changes similar to those observed upon restoration of TET2 expression. Finally, ascorbate treatment enhanced the efficacy of PARP inhibitors, increased the sensitivity of AML cells to DNA damage, and suppressed leukemogenesis in vitro.110 Together, these studies suggest that vitamin C supplementation might be important to maximize residual TET tumor suppressor function, especially because patients with hematological diseases often display low ascorbate levels.111 Whether vitamin C deficiency heightens the risk of hematopoietic malignancies remains to be determined.
Conclusion
Recent large-scale sequencing studies have shown that a large number of patients harbor TET2 LOF mutations in both myeloid and lymphoid malignancies as well as in the premalignant condition CHIP/ARCH. The bulk of the evidence indicates that TET2 inactivation is an early event in the initiation of hematopoietic malignancies, although additional hits are necessary for further tumor progression. These second hits are largely secondary oncogenic mutations in signaling molecules, including FLT3, RAS, and RHOA, although recent studies indicate that cytokines and other inflammatory signals or vitamin C deficiency can also promote cancer progression. Moreover, even in the absence of TET coding region mutations, aberrant expression or function of diverse metabolic enzymes can diminish TET function.
Despite these advances, the underlying mechanisms remain unknown. Why do TET2 and DNMT3A mutations in HSCs induce clonal expansion? Why do TET2 mutations, when combined with mutations in signaling enzymes, such as FLT3 and RHOA, cooperatively induce and maintain hematopoietic malignancies? Why are TET2 mutations only found in certain types of lymphoid malignancies? Why are TET2 and IDH1/2 mutations mutually exclusive in myeloid neoplasms but not in T-cell lymphomas?
From a clinical perspective, it might be useful to modulate TET function in patients with high-dose vitamin C or manipulations that increase αKG and/or decrease 2HG. It might also be worthwhile to develop diagnostic pipelines that include measurements of 5hmC in cancer samples, so as to be able to add the dimension of high or low TET activity in the cancer to the personalized evaluation of each cancer patient. This would accomplish the important goal of classifying hematopoietic cancers by the criterion of what mutations co-occur or are mutually exclusive with global TET LOF, rather than simply mutations in TET2.
Acknowledgments
The authors thank Xiaojun “Max” Xu and Chen “Alice” Zhang for assistance with visualization of the TET2-DNA structure.
This work was supported by National Institutes of Health grants R35 CA210043 (National Cancer Institute), AI109842 and AI128589 (National Institute of Allergy and Infectious Diseases), and Translation Research Project grants 6187-12 and 6464-15 from the Leukemia and Lymphoma Society (A.R.). C.-W.J.L. was supported by an Independent Investigator Fund (Kyowa Hakko Kirin Co./La Jolla Institute) and an Irvington Postdoctoral Fellowship from the Cancer Research Institute.
Authorship
Contribution: C.-W.J.L., H.Y., and A.R. wrote the review.
Conflict-of-interest disclosure: A.R. is a member of the Scientific Advisory Board of Cambridge Epigenetix. The remaining authors declare no competing financial interests.
Correspondence: Anjana Rao, La Jolla Institute for Immunology, 9420 Athena Cir, La Jolla, CA 92037; e-mail: arao@lji.org; and Chan-Wang J. Lio, La Jolla Institute for Immunology, 9420 Athena Cir, La Jolla, CA 92037; e-mail: lio@lji.org.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal