

Key Points
Shear stress–induced release of RNA from endothelial cells is crucial for initiation of arteriogenesis by controlling mechanotransduction.
Extracellular RNA is essential for VWF release from endothelial cells initiating the inflammatory process driving arteriogenesis.

Abstract
Fluid shear stress in the vasculature is the driving force for natural bypass growth, a fundamental endogenous mechanism to counteract the detrimental consequences of vascular occlusive disease, such as stroke or myocardial infarction. This process, referred to as “arteriogenesis,” relies on local recruitment of leukocytes, which supply growth factors to preexisting collateral arterioles enabling them to grow. Although several mechanosensing proteins have been identified, the series of mechanotransduction events resulting in local leukocyte recruitment is not understood. In a mouse model of arteriogenesis (femoral artery ligation), we found that endothelial cells release RNA in response to increased fluid shear stress and that administration of RNase inhibitor blocking plasma RNases improved perfusion recovery. In contrast, treatment with bovine pancreatic RNase A or human recombinant RNase1 interfered with leukocyte recruitment and collateral artery growth. Our results indicated that extracellular RNA (eRNA) regulated leukocyte recruitment by engaging vascular endothelial growth factor receptor 2 (VEGFR2), which was confirmed by intravital microscopic studies in a murine cremaster model of inflammation. Moreover, we found that release of von Willebrand factor (VWF) as a result of shear stress is dependent on VEGFR2. Blocking VEGFR2, RNase application, or VWF deficiency interfered with platelet–neutrophil aggregate formation, which is essential for initiating the inflammatory process in arteriogenesis. Taken together, the results show that eRNA is released from endothelial cells in response to shear stress. We demonstrate this extracellular nucleic acid as a critical mediator of mechanotransduction by inducing the liberation of VWF, thereby initiating the multistep inflammatory process responsible for arteriogenesis.
Introduction
Vascular occlusive diseases are the leading cause of morbidity and mortality worldwide. Interestingly, the human body has an endogenous mechanism that can compensate for the loss of an artery. Preexisting arteriolar connections can grow to form a natural bypass, thereby substituting for the occluded vessel.1 Accordingly, much effort has been spent to unravel the molecular mechanisms of this process, called “arteriogenesis,” to enable clinicians to promote this process in patients suffering from myocardial infarction, stroke, or peripheral artery disease (PAD). If successful, this would lead to a substitution for invasive treatments, such as percutaneous luminal angioplasty, bypass surgery, or even heart transplantation.
Intensive research over the last years revealed that arteriogenesis relies on a local inflammatory process that is triggered by fluid shear stress.2 Upon narrowing or occlusion of a feeding artery, blood flow is redirected into preexisting arteriolar connections. In these vessels, the subsequently increased fluid shear stress causes an activation of the endothelium, followed by an inflammatory cascade initiated by platelet receptor glycoprotein 1bα (GPIbα) and platelet–neutrophil aggregate (PNA) formation.3-5 Finally, perivascular-recruited macrophages promote collateral artery growth by a cytokine burst to restore blood flow.6
A mechanosensory complex consisting of platelet endothelial cell adhesion molecule-1 (PECAM-1), the cell–cell adhesion receptor vascular endothelial cell (EC) cadherin, and vascular endothelial growth factor receptor 2 (VEGFR2) has been identified by in vitro studies, and results were confirmed in aortas of mice at sites of disturbed blood flow prone to leukocyte recruitment and atherosclerosis.7 However, the mechanisms of mechanosensing of ECs are still incompletely understood.8 Other investigators have shown that, in response to shear stress, ECs release ATP, which activates the G proteins Gq- and G11-coupled purinergic P2Y2 receptors, which are relevant for tyrosine phosphorylation of PECAM-1.9 Subsequently, PECAM-1, which forms a complex with vascular EC cadherin, recruits VEGFR2. Although the direct blockade of VEGFR2 or antibodies directed against vascular endothelial growth factor A (VEGFA) strongly interferes with arteriogenesis, the process is only marginally, if at all, promoted by additional doses of VEGFA (for an overview see Jazwa et al10 ), because endogenous VEGFA levels are not rate limiting in arteriogenesis.11 However, for proper arteriogenesis to occur, enhanced VEGFR2 signaling is required. This is mediated by the nontyrosine kinase coreceptor neuropilin 1 (NRP-1),12 which is capable of binding the VEGRA165 isoform of VEGFA, thereby increasing the local plasma membrane concentration of VEGFA and promoting its binding to VEGFR2.13-15 Interestingly, recent in vitro studies from our group pointed to a role for heparin or extracellular RNA (eRNA) in NRP-1–mediated activation of VEGFR2.16
Nucleic acids, which can be released upon cellular damage, are not inert molecules but trigger diverse biological reactions. For example, they are involved in innate immune reactions related to bacterial killing by neutrophil extracellular traps,17 as well as thrombus and edema formation.18,19 Recently, we were able to demonstrate that eRNA promotes leukocyte recruitment and extravasation in a murine cremaster model of inflammation, identifying eRNA as a proinflammatory factor.20 Arteriogenesis is a matter of innate immunity that involves recruitment of neutrophils, followed by mast cell activation and recruitment of T cells and macrophages.4 Whether eRNA plays a role in arteriogenesis has never been investigated, and it was the topic of the present study.
Materials and methods
Quantification of eRNA and LDH in vitro
Bovine aortic ECs (BAECs) were isolated from bovine aorta, as described,21 and cultured in Dulbecco’s modified Eagle medium containing 10% (volume-to-volume ratio) fetal calf serum. To determine the release of cellular RNA, confluent BAECs were washed twice with phosphate buffered saline (pH 7.4) and maintained under static conditions or exposed to shear stress (12 dyn/cm2) in fetal calf serum–free medium for 60 minutes in a temperature-controlled cone-plate viscosimeter, as previously described.22 Cell supernatants were removed and centrifuged for 5 minutes at 200g to remove cells and cell debris. RNA was isolated using a Master Pure RNA purification kit (Epicentre Biotechnologies, Madison, WI) and quantified using Quant-iT Technology (Invitrogen). Lactate dehydrogenase (LDH) was determined in cell supernatants by measuring its activity using a detection kit (Roche Diagnostics, Basel, Switzerland).
Animals and treatments
All experiments were performed in strict accordance with the German animal legislation guidelines and were approved by the Bavarian Animal Care and Use Committee. Mice were housed in a temperature-controlled room on a 12-hour light-dark cycle and were fed a standard laboratory diet. Our investigations were performed with wild-type C57BL/6J mice (Charles River Laboratories, Sulzfeld, Germany), which also served as controls for our studies on intercellular adhesion molecule 1 (ICAM-1)–deficient mice (Icam1tm1Jcgr),23 which were a generous gift from C. Scheiermann (Walter Brendel Centre of Experimental Medicine), and von Willebrand factor (VWF)–deficient mice (B6.129S2-Vwftm1Wgr/J).24 Because C57BL/6J mice show a high spontaneous blood flow recovery rate, for RNase inhibitor treatment we used SV129 mice (Charles River Laboratories), which show a relatively reduced perfusion recovery.25
C57BL/6J mice were treated IV with bovine RNase A (Thermo Fisher Scientific, Waltham, MA), active or inactive human RNase1 (both manufactured by ProteoGenix, Schiltigheim, France), or DNase (Promega, Madison, WI), at a dose of 50 µg/kg, dissolved in saline starting 30 minutes before femoral artery ligation (FAL) and then every other day until the end of the experiment. To inhibit VEGFR2, mice were treated intraperitoneally with semaxanib (Hycultec, Beutelsbach, Germany), 25 mg/kg per day dissolved in dimethyl sulfoxide (DMSO), starting 1 day before the surgical procedure. The highly selective blocking antibody of VEGFR2, DC101 (Bio X Cell, West Lebanon, NH), was administered equally to semaxanib, at a dose of 20 mg/kg dissolved in saline. SV129 mice were treated IV with 20 U RNase inhibitor (Thermo Fisher Scientific) dissolved in saline immediately after ligation and then daily until day 3 after ligation. To block mast cell degranulation, mice were treated intraperitoneally with cromolyn (Sigma-Aldrich, St. Louis, MO), 20 mg/kg per day dissolved in DMSO, starting 3 days before ligation. The control groups of all experimental set-ups were treated with the appropriate solvent.
Additional methods
For a detailed description of FAL, Laser-Doppler perfusion measurements, tissue sampling, histology, immunohistology, flow cytometry analyses, white blood cell counts, and intravital microscopy of the cremaster muscle please see supplemental Material and methods (available on the Blood Web site).
Statistical analyses
Statistical analyses were calculated with GraphPad Prism 6 (GraphPad Software, La Jolla, CA). Data are mean ± standard error of the mean (SEM). Statistical analyses were conducted as indicated in the figure legends. Results were considered statistically significant at P < .05.
Results
Fluid shear stress induces the release of eRNA
Collateral artery growth is locally induced by increased fluid shear stress in arterioles bypassing a stenosed vessel. Because arteriogenesis is not associated with cell damage at sites of vessel growth, we wanted to investigate whether fluid shear stress might trigger the release of RNA from ECs. Indeed, the application of shear stress to primary ECs in culture resulted in significantly increased levels of eRNA in cell supernatant (Figure 1A). Because increased eRNA levels were not associated with increased LDH activity (Figure 1B), these data indicate that the eRNA was liberated from ECs as a result active release and not cell damage.
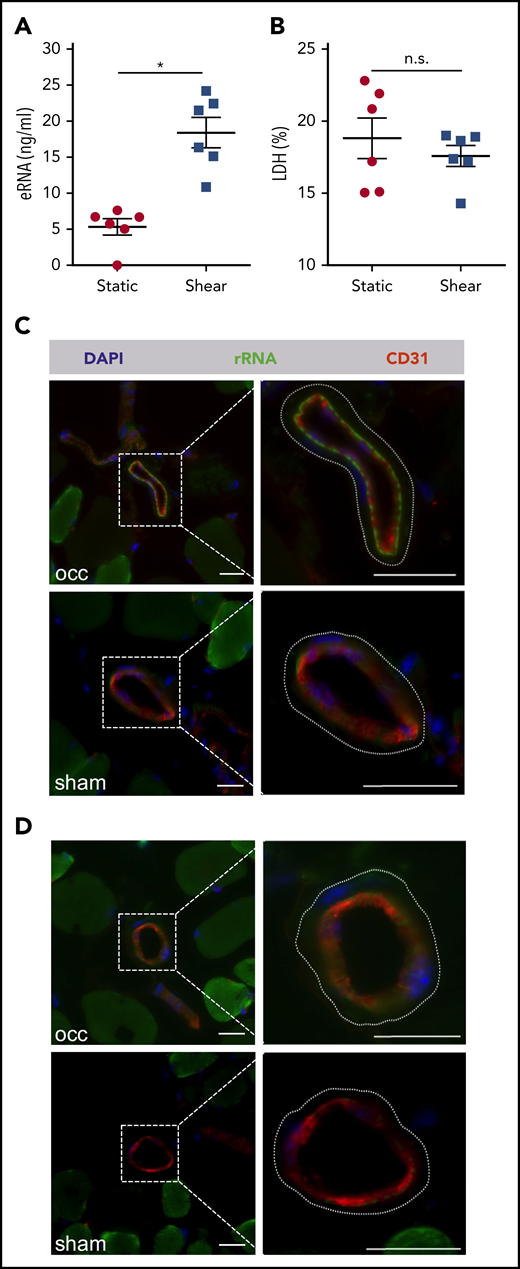
In vitro and in vivo analysis of eRNA release. (A) eRNA levels were measured in supernatant of BAECs, with or without shear conditions of 12 dyn/cm2 for 60 minutes. (B) The levels of LDH (expressed as percentage of total) were measured in the same supernatants of the BAECs, exposed to shear or under static conditions. Data are mean ± SEM; n = 6 per group. (C-D) Representative fluorescent immunohistological pictures of collaterals of mice 30 minutes after induction of arteriogenesis (occ) or sham operation. To visualize collaterals, tissue samples were stained with an antibody against CD31 (red) depicting ECs. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole. The dashed lines in the magnified images (right panels) delineate the outer border of the vessels of the smooth muscle cells. A specific signal for the IV applied fluorescently labeled antibody against ribosomal RNA (green) was found at the abluminal side of ECs of growing collaterals (occ, upper panel), but not of resting collaterals (sham, lower panel) (C), or control mice (D). Scale bars, 20 μm. *P < .05, unpaired Student t test. n.s., not significant.
In vitro and in vivo analysis of eRNA release. (A) eRNA levels were measured in supernatant of BAECs, with or without shear conditions of 12 dyn/cm2 for 60 minutes. (B) The levels of LDH (expressed as percentage of total) were measured in the same supernatants of the BAECs, exposed to shear or under static conditions. Data are mean ± SEM; n = 6 per group. (C-D) Representative fluorescent immunohistological pictures of collaterals of mice 30 minutes after induction of arteriogenesis (occ) or sham operation. To visualize collaterals, tissue samples were stained with an antibody against CD31 (red) depicting ECs. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole. The dashed lines in the magnified images (right panels) delineate the outer border of the vessels of the smooth muscle cells. A specific signal for the IV applied fluorescently labeled antibody against ribosomal RNA (green) was found at the abluminal side of ECs of growing collaterals (occ, upper panel), but not of resting collaterals (sham, lower panel) (C), or control mice (D). Scale bars, 20 μm. *P < .05, unpaired Student t test. n.s., not significant.
To investigate whether fluid shear stress might also be associated with release of eRNA in vivo, a fluorescently labeled antibody directed against ribosomal RNA was administered IV immediately after induction of arteriogenesis by FAL, making use of the general property of antibodies not to enter cells. Immunohistological analyses of cross-sections of adductor muscles containing growing collaterals isolated 30 minutes after the experimental procedure revealed decoration of the EC layer with eRNA at its basolateral surface, an observation that was not made in resting collaterals from the contralateral sham-operated side (Figure 1C) or from control-treated animals (Figure 1D).
RNase, but not DNase or inactive RNase, treatment interferes with arteriogenesis
To investigate whether extracellular nucleic acids might play a role in arteriogenesis, mice were treated with bovine RNase A or DNase using the FAL model. Following induction of arteriogenesis, RNase A but not DNase treatment provoked a significant reduction in perfusion recovery in comparison with saline-treated control mice (Figure 2A). In this context, it is important to mention that treatment of animals with RNase has no toxic side effect (even when overdosed 20-fold), as demonstrated by our previous in vivo studies.26,27 To examine whether the reduction in perfusion recovery was due to eRNA degradation by RNase or to an unknown function of RNase, mice were treated with recombinant human active or inactive RNase1 (which is the counterpart of bovine RNase A), respectively. Again, perfusion recovery was reduced in mice treated with active RNase1 but not in mice treated with inactive RNase1 (Figure 2B). Moreover, our histological analyses revealed a significant reduction in the luminal diameter of growing collaterals in mice treated with RNase A or active RNase1 but not in mice treated with inactive RNase1 (Figure 2C-D).
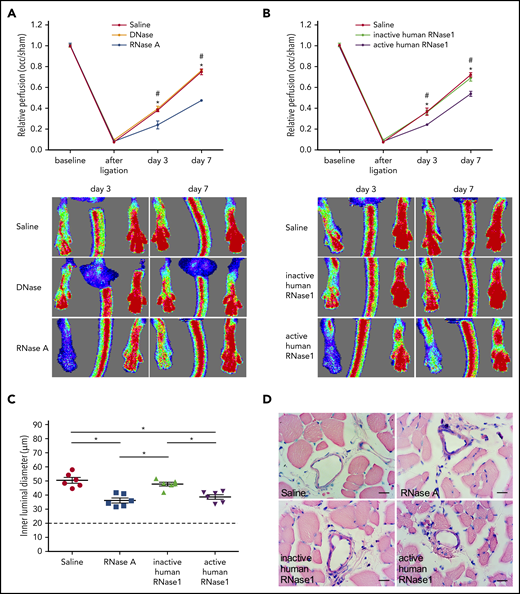
RNase treatment decreases perfusion recovery and vessel growth. (A-B) Laser Doppler perfusion measurements (upper panels) and corresponding flux images (lower panels) of mice treated as indicated. The perfusion was calculated using occluded/sham (right to left) ratios before the surgical procedure, immediately after the surgical procedure, and at days 3 and 7 after surgery (n = 6). Representative flux images are shown for days 3 and 7. (C) The dot plots show the inner luminal diameter of mice treated with saline, RNase A, or recombinant inactive or active human RNase1 7 days after the surgical procedure (n = 6 per group). Data are mean ± SEM. The dashed horizontal line indicates the mean sham value. (D) Representative photomicrographs of Giemsa-stained tissue samples. Scale bars, 20 µm. *,#P < .05; *saline vs RNase A, #DNase vs RNase A, *saline vs active human RNase1, #inactive human RNase1 vs active human RNase1, 2-way analysis of variance (ANOVA) with the Bonferroni multiple-comparison test (A-B) and 1-way ANOVA with the Bonferroni multiple-comparison test (C).
RNase treatment decreases perfusion recovery and vessel growth. (A-B) Laser Doppler perfusion measurements (upper panels) and corresponding flux images (lower panels) of mice treated as indicated. The perfusion was calculated using occluded/sham (right to left) ratios before the surgical procedure, immediately after the surgical procedure, and at days 3 and 7 after surgery (n = 6). Representative flux images are shown for days 3 and 7. (C) The dot plots show the inner luminal diameter of mice treated with saline, RNase A, or recombinant inactive or active human RNase1 7 days after the surgical procedure (n = 6 per group). Data are mean ± SEM. The dashed horizontal line indicates the mean sham value. (D) Representative photomicrographs of Giemsa-stained tissue samples. Scale bars, 20 µm. *,#P < .05; *saline vs RNase A, #DNase vs RNase A, *saline vs active human RNase1, #inactive human RNase1 vs active human RNase1, 2-way analysis of variance (ANOVA) with the Bonferroni multiple-comparison test (A-B) and 1-way ANOVA with the Bonferroni multiple-comparison test (C).
Because exogenously administered RNA is rapidly degraded by endogenous RNase in blood28 and is suitable for acute, but not chronic, in vivo experiments, such as arteriogenesis20 (see "Discussion"), mice were treated with RNase inhibitor to fully block any endogenous RNases to protect eRNA against degradation. In vitro RNase inhibitor showed no signs of toxicity to induce the release of LDH (supplemental Figure 1A-C) at concentrations used for the in vivo experiments (10 U/mL). In vivo, the administration of RNase inhibitor significantly increased perfusion recovery after FAL, suggesting that an increase in eRNA bioavailability improves arteriogenesis (supplemental Figure 1D). Together, these results confer a decisive role for eRNA in the process of fluid shear stress–induced arteriogenesis.
RNase treatment interferes with neutrophil extravasation and mast cell activation
To investigate whether eRNA plays a role in the previously identified pathway of perivascular macrophage accumulation during collateral artery growth,4 we investigated leukocyte recruitment and mast cell activation in the process of arteriogenesis. Compared with control, treatment of mice with RNase A or active RNase1 resulted in a significant reduction in the number of infiltrated neutrophils (CD45+/Gr1+/CD115−) at day 1 following induction of arteriogenesis (Figure 3A-D). This was accompanied by a reduced number of degranulating mast cells in the perivascular space of growing collateral arteries and was comparable with treatment using the mast cell stabilizer cromolyn (Figure 3E). In contrast, administration of inactive RNase1 did not have an influence on neutrophil recruitment or on mast cell degranulation (Figure 3C-E), confirming that eRNA plays a role in neutrophil-related mast cell activation. At day 3 following RNase A treatment, reduced numbers of infiltrated macrophages (CD45+/F4/80+) were seen, in line with the function of activated and degranulated mast cells in the promotion of perivascular accumulation of macrophages (Figure 3F-H). The parallel analyses of blood samples on day 1 and day 3 revealed that RNase A treatment had no influence on the number of circulating leukocyte subsets (Figure 3I-J). This indicates that RNase treatment exclusively affected perivascular leukocyte recruitment but not their mobilization from the bone marrow.
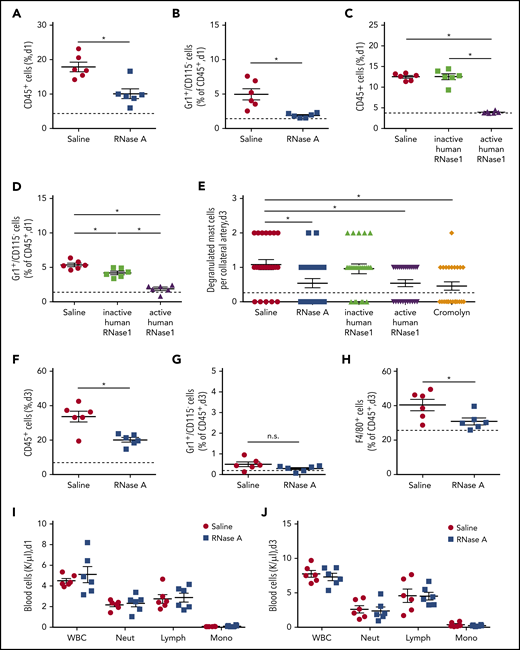
RNase treatment reduces leukocyte infiltration and mast cell degranulation. (A-D,F-H) Quantitative analyses, using flow cytometry, of infiltrated leukocytes in adductor muscles isolated at day 1 or 3 after induction of arteriogenesis via FAL. Scatter plots show the percentages of CD45+ cells (A,C,F), CD45+/Gr1+/CD115− cells (B,D,G), and CD45+/F4/80+ cells (H) in adductor muscles from mice treated with saline or RNase A (A-B,F-H) or from mice treated with saline, recombinant inactive human RNase1, or recombinant active human RNase1 (C-D). Analyses were performed at day 1 (A-D) and day 3 (F-H) after FAL. (E) The scatter plot shows the number of degranulated mast cells in the perivascular space of growing collaterals at day 3 after induction of arteriogenesis in mice treated with saline, RNase A, recombinant active or inactive human RNase1, or the mast cell stabilizer cromolyn, as evaluated on Giemsa-stained tissue sections. Counts of white blood cell (WBC) populations in blood samples drawn from mice at day 1 (I) or day 3 (J) after induction of arteriogenesis by FAL and saline or RNase A treatment. Data are mean ± SEM. n = 6 mice per group (A-D,F-J), n > 10 mice per group (E). The dashed horizontal line in the scatter plots indicates the mean sham value. *P < .05, unpaired Student t test (A-B,F-J), 1-way ANOVA with the Bonferroni multiple-comparison test (C-E). Lymph, lymphocytes; Mono, monocytes; Neut, neutrophils.
RNase treatment reduces leukocyte infiltration and mast cell degranulation. (A-D,F-H) Quantitative analyses, using flow cytometry, of infiltrated leukocytes in adductor muscles isolated at day 1 or 3 after induction of arteriogenesis via FAL. Scatter plots show the percentages of CD45+ cells (A,C,F), CD45+/Gr1+/CD115− cells (B,D,G), and CD45+/F4/80+ cells (H) in adductor muscles from mice treated with saline or RNase A (A-B,F-H) or from mice treated with saline, recombinant inactive human RNase1, or recombinant active human RNase1 (C-D). Analyses were performed at day 1 (A-D) and day 3 (F-H) after FAL. (E) The scatter plot shows the number of degranulated mast cells in the perivascular space of growing collaterals at day 3 after induction of arteriogenesis in mice treated with saline, RNase A, recombinant active or inactive human RNase1, or the mast cell stabilizer cromolyn, as evaluated on Giemsa-stained tissue sections. Counts of white blood cell (WBC) populations in blood samples drawn from mice at day 1 (I) or day 3 (J) after induction of arteriogenesis by FAL and saline or RNase A treatment. Data are mean ± SEM. n = 6 mice per group (A-D,F-J), n > 10 mice per group (E). The dashed horizontal line in the scatter plots indicates the mean sham value. *P < .05, unpaired Student t test (A-B,F-J), 1-way ANOVA with the Bonferroni multiple-comparison test (C-E). Lymph, lymphocytes; Mono, monocytes; Neut, neutrophils.
VWF release from ECs is dependent on eRNA and VEGFR2
It has been shown in vitro and in vivo that VWF is released from ECs as (ultra-)large multimers under conditions of increased shear stress.29,30 VWF provides a major ligand for platelet receptor GPIbα, mediating effective platelet adhesion, particularly under high fluid shear stress.29,31 In addition to other mechanisms, it is well described that VEGFA induces VWF release from endothelial Weibel-Palade bodies (WPBs) by activating the cognate VEGFR2.32 Therefore, we asked whether VEGFR2 is relevant for VWF release under conditions of increased shear stress during arteriogenesis and whether eRNA might play a role in this process.
Administration of the VEGFR2 inhibitor semaxanib (SU5416) significantly interfered with perfusion recovery and collateral artery growth (Figure 4A-D), confirming previous results.10 To investigate whether VWF is released from collateral ECs during the process of arteriogenesis, we again took advantage of the properties of antibodies not to be able to enter cells and administered an IV antibody against VWF 30 minutes prior to tissue sampling. Our immunohistological analyses performed on tissue samples isolated 2 hours after the surgical procedure showed that induction of arteriogenesis resulted in the release of VWF from ECs of growing collateral arteries (Figure 4E-F). This release process was almost completely abolished when mice were pretreated with semaxanib, a VEGFR2-blocking antibody (DC101), or RNase A (Figure 4E-F).
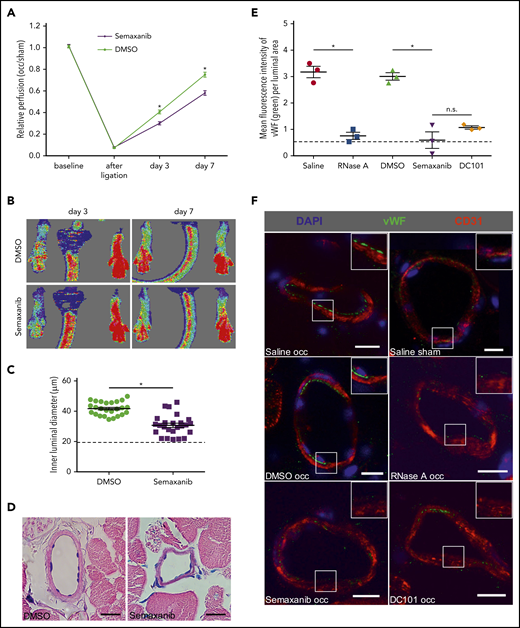
RNase treatment and VEGFR2 blockade interfere with VWF release during arteriogenesis. Laser Doppler perfusion measurements (A) along with corresponding flux images (B) of DMSO (solvent)-treated mice compared with semaxanib-treated mice. The perfusion recovery was calculated by the occluded/sham (right to left) ratio before the surgical procedure, immediately after the surgical procedure, and 3 days and 7 days after ligation. Representative flux images of the Laser Doppler perfusion measurements are shown for day 3 and day 7. The scatter plot shows the inner luminal diameter of mice treated with DMSO or semaxanib 7 days after FAL (C) along with representative Giemsa stains of evaluated tissue samples (D). Scale bars, 20 µm. (E) The scatter plot shows the amount of luminal VWF (as calculated by quantifying the mean green fluorescence intensity per luminal area) in mice treated with saline, DMSO, RNase A, semaxanib, or DC101 (antibody blocking VEGFR2) 2 hours after FAL. (F) Representative immunohistological stains of evaluated collateral arteries. ECs of growing collaterals are strongly decorated with VWF on their luminal surface (green dots; top and middle left panels); this is not visible in the other panels (saline sham, RNase A occ, semaxanib occ, or DC101 occ). Scale bars, 10 µm. Insets show magnifications of collateral arteries in boxes. Data are mean ± SEM. n = 6 per group (A-B), n > 10 per group (C-D), n = 3 per group (E-F). The dashed horizontal line in the scatter plots indicates the mean sham value. *P < .05, 2-way ANOVA with the Bonferroni multiple-comparison test (A), unpaired Student t test (C), 1-way ANOVA with the Bonferroni multiple-comparison test (E).
RNase treatment and VEGFR2 blockade interfere with VWF release during arteriogenesis. Laser Doppler perfusion measurements (A) along with corresponding flux images (B) of DMSO (solvent)-treated mice compared with semaxanib-treated mice. The perfusion recovery was calculated by the occluded/sham (right to left) ratio before the surgical procedure, immediately after the surgical procedure, and 3 days and 7 days after ligation. Representative flux images of the Laser Doppler perfusion measurements are shown for day 3 and day 7. The scatter plot shows the inner luminal diameter of mice treated with DMSO or semaxanib 7 days after FAL (C) along with representative Giemsa stains of evaluated tissue samples (D). Scale bars, 20 µm. (E) The scatter plot shows the amount of luminal VWF (as calculated by quantifying the mean green fluorescence intensity per luminal area) in mice treated with saline, DMSO, RNase A, semaxanib, or DC101 (antibody blocking VEGFR2) 2 hours after FAL. (F) Representative immunohistological stains of evaluated collateral arteries. ECs of growing collaterals are strongly decorated with VWF on their luminal surface (green dots; top and middle left panels); this is not visible in the other panels (saline sham, RNase A occ, semaxanib occ, or DC101 occ). Scale bars, 10 µm. Insets show magnifications of collateral arteries in boxes. Data are mean ± SEM. n = 6 per group (A-B), n > 10 per group (C-D), n = 3 per group (E-F). The dashed horizontal line in the scatter plots indicates the mean sham value. *P < .05, 2-way ANOVA with the Bonferroni multiple-comparison test (A), unpaired Student t test (C), 1-way ANOVA with the Bonferroni multiple-comparison test (E).
eRNA and VEGFR2 play a crucial role in PNA formation
We have previously shown that platelet activation by platelet receptor GPIbα, followed by PNA formation, is a prerequisite for mast cell degranulation in the context of arteriogenesis.4 In the course of the present study, treatment of mice with RNase A or with the VEGFR2 blocker semaxanib, as well as VWF deficiency, significantly interfered with PNA formation. These data indicate that the process of arteriogenesis is dependent on VWF release mediated by activation of VEGFR2 involving eRNA (Figure 5A-C). Under these experimental conditions, the numbers of peripheral blood neutrophils and platelets were not altered (Figure 5D-E), suggesting that the reduced PNA formation in differently treated mice was not due to a reduced bioavailability of 1 of the PNA components.
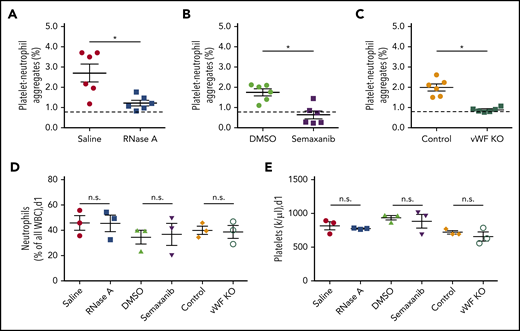
VWF is essential for PNA formation in the process of arteriogenesis. Following induction of arteriogenesis at day 1, whole blood was drawn from wild-type mice treated with saline or RNase A (A) or with DMSO or semaxanib (B) or from VWF-deficient (VWF KO) mice and their respective controls (C) and analyzed using flow cytometry. The scatter plots show the percentage of PNAs relative to the total number of neutrophils. Platelets were detected by a CD41 antibody, and neutrophils were identified by CD11b and Gr-1 antibodies. The dashed horizontal line indicates the mean sham value. For comparison, blood neutrophils (D) and platelets (E) were quantified in wild-type mice treated with saline, RNase A, DMSO, or semaxanib, as well as in VWF-deficient (VWF KO) mice and untreated wild-type mice (Control). Data are mean ± SEM. n = 6 per group (A-C), n = 3 per group (D-E). *P < .05, unpaired Student t test.
VWF is essential for PNA formation in the process of arteriogenesis. Following induction of arteriogenesis at day 1, whole blood was drawn from wild-type mice treated with saline or RNase A (A) or with DMSO or semaxanib (B) or from VWF-deficient (VWF KO) mice and their respective controls (C) and analyzed using flow cytometry. The scatter plots show the percentage of PNAs relative to the total number of neutrophils. Platelets were detected by a CD41 antibody, and neutrophils were identified by CD11b and Gr-1 antibodies. The dashed horizontal line indicates the mean sham value. For comparison, blood neutrophils (D) and platelets (E) were quantified in wild-type mice treated with saline, RNase A, DMSO, or semaxanib, as well as in VWF-deficient (VWF KO) mice and untreated wild-type mice (Control). Data are mean ± SEM. n = 6 per group (A-C), n = 3 per group (D-E). *P < .05, unpaired Student t test.
eRNA initiates mast cell–mediated macrophage recruitment and vascular cell proliferation
The administration of RNase A or semaxanib, as well as a VWF deficiency, in mice also significantly interfered with mast cell degranulation without influencing mast cell recruitment per se (supplemental Figure 2), accounting for an eRNA-induced signaling pathway of mast cell activation. The treatment of mice with RNase A or semaxanib interfered with perivascular M1-polarized (CD68+MRC1−) and M2-polarized (CD68+MRC1+) macrophage accumulation (supplemental Figure 3). Comparable results were obtained with mice that were deficient for ICAM-1 (an endothelial adhesion receptor whose increased expression is dependent on enhanced VEGFR2/NRP-1 signaling33 ), which is upregulated during arteriogenesis34 and is relevant for monocyte adhesion during collateral artery growth,35 as well as in mice that were treated with the mast cell degranulation blocker cromolyn (supplemental Figure 3). These latter results are in line with our previous data showing that mast cell activation/degranulation is essential for perivascular macrophage recruitment in arteriogenesis.4 Moreover, reduced perivascular macrophage accumulation under the same experimental settings was associated with a reduced proliferation of collateral artery ECs and smooth muscle cells after induction of arteriogenesis (supplemental Figure 4). These findings underline the relevance of leukocytes as a source of growth factors and cytokines for the process of arteriogenesis.36,37
eRNA mediates VEGFR2-induced inflammation
To further confirm our data that eRNA is essential for VEGFR2-promoted inflammation and to investigate the relevance of our findings, an acute murine trauma model (cremaster model) of inflammation was included.20 Intrascrotal injection of RNA induced leukocyte adhesion to a comparable level as did tumor necrosis factor α (TNF-α), which was used as a positive control. The blockade of VEGFR2 by semaxanib significantly interfered with eRNA-induced, but not with TNF-α–promoted, leukocyte adhesion (Figure 6A). Interestingly, when TNF-α was applied the rolling flux fraction was reduced, although not significantly. However, due to as yet unknown reasons this was not the case when eRNA was administered (Figure 6B). Together, our data confirm that eRNA provides its proinflammatory action via VEGFR2 and highlight the relevance of our findings for shear stress–induced arteriogenesis, as well as for acute and chronic inflammatory processes in general. However, our data also point to VEGFR2-independent mechanisms of inflammation because TNF-α–induced leukocyte recruitment was not influenced by VEGFR2 blockage.
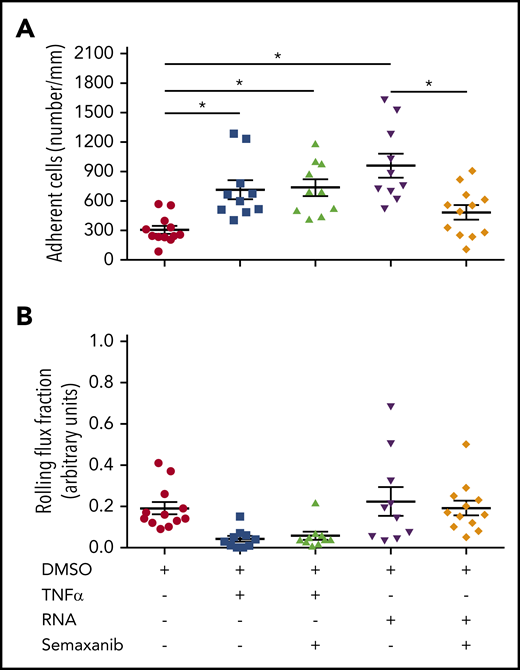
eRNA activates VEGFR2 in a murine cremaster model of inflammation activation. In a cremaster model of inflammation, adherent leukocytes (A) and the rolling flux fraction (B) were investigated using intravital microscopy in venules of mice pretreated with semaxanib blocking VEGFR2 or solvent (DMSO) and stimulated with TNF-α or RNA. Data are mean ± SEM. n > 10 per group. *P < .05, 1-way ANOVA with the Bonferroni multiple-comparison test.
eRNA activates VEGFR2 in a murine cremaster model of inflammation activation. In a cremaster model of inflammation, adherent leukocytes (A) and the rolling flux fraction (B) were investigated using intravital microscopy in venules of mice pretreated with semaxanib blocking VEGFR2 or solvent (DMSO) and stimulated with TNF-α or RNA. Data are mean ± SEM. n > 10 per group. *P < .05, 1-way ANOVA with the Bonferroni multiple-comparison test.
Discussion
Arteriogenesis is a fluid shear stress–triggered process that is strongly dependent on local leukocyte recruitment. Previous studies identified several signaling processes that are involved in transducing shear stress–mediated effects. However, how leukocytes are locally recruited to sites of sterile inflammation is not clear. In the present study, we identified several missing links in translating fluid shear stress, the triggering force for arteriogenesis, to leukocyte recruitment, thereby promoting collateral artery growth. This allows us to plot the circle of complex mechanisms of cellular and molecular chain reactions in arteriogenesis (Figure 7). In particular, we show that shear stress triggers the release of RNA from preexisting collateral arteries. eRNA, in turn, acts as a mechanotransducer provoking the release of VWF, which subsequently initiates the local inflammatory cascade mediating collateral artery growth.
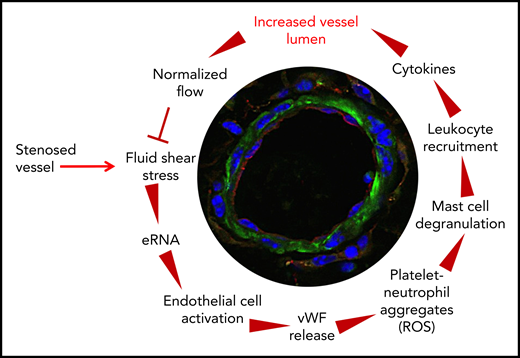
The circle of arteriogenesis. Upon occlusion of a feeding artery, blood flow is redirected in preexisting collaterals circumventing the stenosed artery. Because of the increased blood flow, collaterals experience increased shear stress, which results in release of RNA from ECs. eRNA acts as mechanotransducer provoking EC activation. Activated ECs release VWF from their WPBs, which activates platelets to form PNAs. These, in turn, are essential for mast cell activation, which is a prerequisite for the recruitment of leukocytes that boost vascular cell proliferation and, hence, vessel growth by supplying growth factors and cytokines. When collaterals reached a critical size, allowing them to substitute for the function of the occluded artery, blood flow is normalized, and the collaterals cease to grow.
The circle of arteriogenesis. Upon occlusion of a feeding artery, blood flow is redirected in preexisting collaterals circumventing the stenosed artery. Because of the increased blood flow, collaterals experience increased shear stress, which results in release of RNA from ECs. eRNA acts as mechanotransducer provoking EC activation. Activated ECs release VWF from their WPBs, which activates platelets to form PNAs. These, in turn, are essential for mast cell activation, which is a prerequisite for the recruitment of leukocytes that boost vascular cell proliferation and, hence, vessel growth by supplying growth factors and cytokines. When collaterals reached a critical size, allowing them to substitute for the function of the occluded artery, blood flow is normalized, and the collaterals cease to grow.
Nucleic acids and, in particular, RNA, as a result of its extranucleic localization, have been described to be released from cells as a result of damage.38 However, nucleic acids can be actively released from cells, as shown for neutrophils forming neutrophil extracellular traps.17,39 Here, we show that fluid shear stress provides a trigger for the release of eRNA from ECs, although the exact mechanism of liberation remains to be elucidated. Depending on the cell type and the agonist used, eRNA can be released as free RNA or in association with microvesicles40,41 (S.F. and K.T.P., unpublished observations). In the case of arteriogenesis, it appears that mainly free ribosomal RNA is released as eRNA from ECs, because this tissue-associated eRNA was stained by a specific antibody or became readily degraded by administered RNase. In contrast, microvesicle-entrapped eRNA would not be recognized by an IV-administered antibody or RNase.40 The identified proinflammatory mechanisms that allow eRNA to act as a danger-associated molecular pattern apply for shear stress–induced arteriogenesis, as well as for acute and chronic inflammatory processes in general, as documented previously.41,42 In fact, ribosomal RNA makes up >80% of cellular RNA and constitutes the majority of RNA released, once a cell becomes stressed or damaged. In earlier reports, eRNA has been described as a damaging factor in cardiovascular pathologies, and it was demonstrated that administration of RNase1 interfered with the adverse effects of eRNA.42,43 In sharp contrast, the present study strongly indicates that endogenous eRNA serves as a potent mechanotransducing and inflammatory factor that is necessary for effective natural bypass growth. Administration of active RNases (bovine pancreatic RNase A or human recombinant RNase1), but not the inactive endonuclease or DNase, strongly reduced the process of collateral artery growth, pointing to eRNA as a key factor in arteriogenesis. Although one might expect RNase1 to have cytotoxic side effects, RNase1 has been demonstrated to exert its activity exclusively outside of cells.44 So far, no distinct receptor for RNase1 has been identified,45 and despite the fact that the enzyme might enter cells by endocytosis,46 it is rapidly inactivated by the high cytosolic concentrations of RNase inhibitor.47,48 In accordance with the function of eRNA as a driving force in arteriogenesis and the counteracting activity of vascular RNases, the bioavailability of eRNA and, hence, the perfusion recovery could be enhanced by administration of RNase inhibitor in the mouse FAL model to abrogate the endogenous RNases and to prevent rapid eRNA degradation.
In a murine cremaster model of inflammation, we recently demonstrated that eRNA promotes leukocyte recruitment and extravasation as strongly as does TNF-α.20 The process of arteriogenesis follows the different steps of a sterile inflammatory pathway. Using the same hindlimb model of arteriogenesis as used here, we demonstrated that arteriogenesis is dependent on activation of the platelet receptor GPIbα,3 which results in PNA formation causing extracellular superoxide anion and, hence, reactive oxygen species formation of neutrophils. After extravasation, neutrophil-derived reactive oxygen species activate mast cells, which, in turn, recruit monocytes, which, in the form of perivascular macrophages, promote arteriogenesis by delivering growth factors and cytokines.4 The ligands for GPIbα are manifold,49 and the question about which factor activates the receptor in response to fluid shear stress, resulting in arteriogenesis, remained open. Here, we show that deficiency of VWF strikingly interfered with PNA formation without affecting the number of platelets or neutrophils in peripheral blood, identifying VWF as a relevant factor for GPIbα activation.
Interestingly, PNA formation in the arteriogenesis model did not create any prothrombotic situation that would be detrimental for the blood flow reconstitution in growing collaterals. We did not observe any thrombus formation in the growing collaterals, possibly because of the high shear rate–dependent increased production of nitric oxide and prostacyclin in these vessels, thereby blocking platelet aggregation and subsequent fibrin formation.5,30,31,50-54 Moreover, under these conditions, ultralarge VWF multimers may become degraded by ADAMTS13,30,31,52 resulting in a transient, but not firm, adhesion of platelets to the endothelium of growing collaterals.3 Yet, further investigations are needed to address this issue in more detail.
Shear stress has been closely correlated with VWF release and action.29,31 Again, a variety of agonists, among them VEGF and TNF-α, have been described to induce WPB exocytosis; however, the mechanisms relevant for shear stress–triggered release of VWF from WPBs remained to be elucidated.55-57 Our results revealed that blocking VEGFR2 interfered with VWF release and subsequent PNA formation, mast cell activation, leukocyte recruitment, and, finally, the process of arteriogenesis. Similar effects were seen upon treatment of mice with RNase A. It has previously been shown that mutation of Tyr1175 in the C terminus of VEGFR2 abolished VWF release from ECs in vitro.32 For the process of arteriogenesis, it was demonstrated that NRP-1, the coreceptor of VEGFA, which is essential for enhancing VEGFR2 signaling, is required for VEGFR2 Tyr1175 phosphorylation, thereby playing a profound role in the process of collateral artery growth.12 Moreover, we have shown in vitro that eRNA initiates the binding of VEGFA to NRP-1, thereby increasing the local concentration of VEGFA relevant for proper binding of the cytokine to VEGFR2 and Tyr1175 phosphorylation.16 In the present study, we observed an intense immunostaining for eRNA at the abluminal site of ECs in growing collaterals. Although we cannot exclude that eRNA was removed during the process of perfusion fixation of the tissue or as a result of degradation of eRNA by RNases in blood circulation, our data are in line with immunocytochemical data localizing VEGFA at the abluminal plasma membrane of ECs in vivo58 and corresponding to the anticipated distribution of VEGFRs and VEGFR2 activity.59,60
To follow these leukocyte-dependent reactions and to provide proof-of-principle data in the context of arteriogenesis, experiments were performed with eRNA in the acute murine cremaster vasculature model of acute inflammation, because in vivo administration of eRNA would not be suitable for chronic experiments, such as the FAL model. Our results showed that blocking VEGFR2 strictly interferes with eRNA-induced, but not with TNF-α–induced, leukocyte recruitment, confirming that the inflammatory properties of eRNA are mediated by VEGFR2. Nevertheless, eRNA is also likely to be involved in TNF-α–related inflammation. We have previously demonstrated that the increased bioavailability of TNF-α in arteriogenesis is dependent on mast cell degranulation,4 and our current results suggest that the relevant signal transduction cascade resulting in mast cell degranulation is initiated by eRNA. Finally, it has been described that eRNA liberates TNF-α from macrophages in a TNF-α–converting enzyme–dependent manner.41 However, eRNA signaling seems to have an even broader relevance for inflammatory processes. The facts that P-selectin is concomitantly released with VWF from WPBs and translocated to the cell surface,61-64 and that increased expression of ICAM-1 is dependent on enhanced VEGFR2 signaling mediated by engagement of NRP-133 suggest that eRNA might also have a function in rolling and firm adhesion of leukocytes. These assumptions are in line with our previous findings on RNA stimulation of cremaster muscle vessels.20
Taken together, our work identified eRNA, released as a result of shear stress, as a relevant factor mediating mechanotransduction and, thereby, initiating the inflammatory response triggering natural bypass growth. However, the discussed mechanisms of eRNA-mediated inflammation are likely to apply to shear stress–driven leukocyte recruitment, as well as to other chronic and acute types of inflammation in which RNA is released because of cell damage, conferring a major role for eRNA in innate immune reactions.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank C. Csapo for technical assistance.
This work was supported by the Lehre@LMU program from the Ludwig Maximilian University of Munich.
Authorship
Contribution: M.L., E.C.K., J.-I.B., T.G., and T.L. performed in vivo measurements; M.L., E.C.K., S.M., K.K., and K.T. performed histological analyses; M.L. and E.C.K. performed FACS analyses; S.S., S.F., and I.F. performed in vitro analyses; I.F., A.M.R., M.S., and K.T.P. participated in scientific discussions and drafted and revised the manuscript; and E.D. designed the experiments, analyzed the data, and wrote the manuscript.
Conflict-of interest disclosure: The authors declare no competing financial interests.
Correspondence: Elisabeth Deindl, Walter Brendel Centre of Experimental Medicine, Marchioninistr 15, 81377 Munich, Germany; e-mail: elisabeth.deindl@med.uni-muenchen.de.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal