

TO THE EDITOR:
Since the discovery of circulating cell-free DNA (cfDNA) in the plasma of pregnant women in the late 1990s,1,2 its potential for prenatal diagnosis has been the focus of intensive technological innovation. Screening for chromosomal abnormalities is now introduced in several countries, including the United Kingdom.3 However, the so-called combined test (which utilizes ultrasound scanning to measure fetal nuchal translucency, maternal age, and blood tests to measure pregnancy-associated plasma protein A and free β-human chorionic gonadotrophin) remains the first-stage test. Women at high risk of carrying an affected baby are offered noninvasive prenatal testing (NIPT), and if this returns a positive result, they are given the option of confirmatory invasive testing by amniocentesis.
Technologies detecting dominant de novo mutations or dominant mutations of paternal origin from cfDNA have been previously described.4 For recessive disorders (eg, cystic fibrosis) where the patient is a compound heterozygote, the same technology can be applied, because the mutations in both parents are different. Where mutations are the same, methods utilizing the linkage of disease-causing mutations to paternal single-nucleotide polymorphisms have been proposed.5,6 However, this requires testing a previously born child for linkage to paternal single-nucleotide polymorphisms, and it is subject to errors due to recombination events. An alternative approach avoiding the need for complex family workup would be to measure small allelic imbalances caused by fetal DNA in the maternal circulation. However, this requires precise allele quantification and, so far, rapid NIPT from cfDNA for conditions such as sickle-cell anemia (SCA), where the mother and father carry the same mutation, remains elusive.
SCA is an autosomal recessive disease characterized by a single base-pair substitution in the β globin gene. Due to the protective effect of the mutation against malaria, carrier frequencies in sub-Saharan Africa are ≥20%. Over 224 200 infants are born annually with SCA worldwide,7 including at least 1000 in the United States, making SCA the most common monogenic disease indication for invasive prenatal testing (IPT) in high-income countries. Invasive prenatal diagnosis by amniocentesis or chorionic villus sampling is costly and carries a 1% to 2% risk of miscarriage. Only 1 study so far has reported the use of cfDNA for the detection of allelic imbalance in SCA using digital polymerase chain reaction (dPCR),8 and, to our knowledge, no clinical services currently offer the test anywhere in the world.
Here, we develop and validate a highly sensitive and specific next-generation sequencing approach for the noninvasive prenatal diagnosis of SCA. Our methodology does not require the paternal genotype and combines optimized PCR of the affected locus (including sequencing and PCR error correction) with precise estimation of the fetal cell fraction and different internal controls (Figure 1).9 The raw assay data are analyzed using a bespoke statistical methodology (Figure 2),10,-12 which estimates the fetal disease status (see supplemental Methods, available on the Blood Web site). The method is also applicable to other autosomal recessive and autosomal dominant diseases.
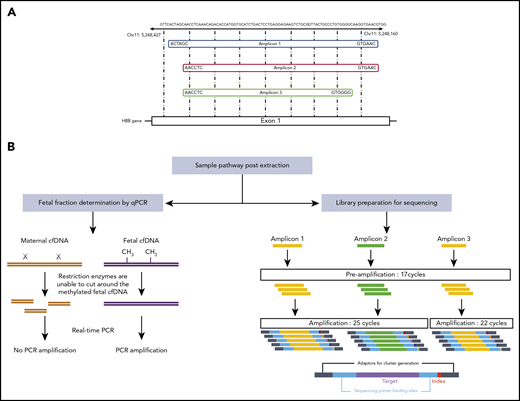
Overview of next-generation sequencing. (A) Three separate primer pairs were designed to generate different amplicons that cover the HbS mutation site. (B) Fetal fractions were determined by the RASSF1A promoter methylation status, as described previously.9 The amount of fetal DNA was assessed by comparing the levels of amplification seen following methylation-sensitive restriction digests with amplification in corresponding undigested samples. For library preparation, single-plex PCRs were performed using 3 different primer pairs to mitigate the effects of any PCR biases that may occur with any individual primer pairs. Small primers were used in an initial limited 17-cycle PCR to preserve the wild-type to sickle allele ratio of the template. A second amplification was performed using longer primers that contained the relevant adapters, primer binding sites, and barcodes necessary for sequencing. As amplicon 3 consistently gave slightly higher yields than amplicons 1 and 2, only 22 cycles were required to generate enough library for sequencing, whereas 25 cycles were required for amplicons 1 and 2. Chr, chromosome.
Overview of next-generation sequencing. (A) Three separate primer pairs were designed to generate different amplicons that cover the HbS mutation site. (B) Fetal fractions were determined by the RASSF1A promoter methylation status, as described previously.9 The amount of fetal DNA was assessed by comparing the levels of amplification seen following methylation-sensitive restriction digests with amplification in corresponding undigested samples. For library preparation, single-plex PCRs were performed using 3 different primer pairs to mitigate the effects of any PCR biases that may occur with any individual primer pairs. Small primers were used in an initial limited 17-cycle PCR to preserve the wild-type to sickle allele ratio of the template. A second amplification was performed using longer primers that contained the relevant adapters, primer binding sites, and barcodes necessary for sequencing. As amplicon 3 consistently gave slightly higher yields than amplicons 1 and 2, only 22 cycles were required to generate enough library for sequencing, whereas 25 cycles were required for amplicons 1 and 2. Chr, chromosome.
![Overview of the statistical analysis. (A) For each amplicon i, we model the expected fraction of reads harboring the HbS allele in the total cfDNA isolated from the mother as a mixture of fetal (ψi) and maternal (ϕi) components in proportions determined by the fetal fraction w. The control shares the same expected fraction of mutated reads ϕi with the mother, which helps increase the precision of the estimates. (B) For each case, we estimate the expected fetal (ψ1,ψ2,ψ3) and maternal (ϕ1,(ϕ2,(ϕ3) fractions of mutated reads per amplicon. (C) We also estimate the overall distribution (given the data) of each triplet of expected maternal and fetal fractions presented in panel B. For a carrier mother (AS), the distribution of the maternal fractions is close to 50%, while for a fetus with the disease (SS), the corresponding distribution is shifted to the right of the maternal distribution. The stronger this shift E[P], the more likely it is that the fetus has the disease. (D) Overview of model training. We identified an optimal E[P] threshold equal to 0.62. The variance of the various performance metrics was estimated using the bootstrap. (E) Overview of applying the calibrated model on both the training and test cohorts. (F-H) Overview of model performance with decreasing fetal fraction. A false negative arises at a fetal fraction of 0.5%, while false positives arise at fetal fractions <4%. Four samples (in green) with E[P] scores close to the threshold are also characterized by small fetal fractions (≤0.9%). Statistical analysis was conducted in R10 and Stan.11,12 Details are given in supplemental Methods. TNR, true negative result; TPR, true positive result.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/134/14/10.1182_blood.2019002099/3/m_bloodbld2019002099f2.png?Expires=1767752944&Signature=r-0UD6AGqmxD8Na1bPHPsO4bnPjp4Tev2gcGCwRjuvKqENJmcukb3dA8t6e9tSYoyuYizFHNwsSrPFNYouMpzNc6frUni8Nu7l3RYgDXWiBW6UG3hb2MA56FeV7FghJNIM5QCuG88oKHpZjPu3VQa~gpY5jzf8xLRcZ5IdsDX6BTJIkRtWcxLVH32WUDQsoxtYbEpuK3i13XkI9fqA9tb0xBg5LxEvOpddoMqMxDNlvm4JOnOcWJNWFBJj1KWZFivYO8rWlcazCdZO7Te3ri61iWDXfOc1M2tReCiMiYAC76YWCVO1nzcOkjYm805gDz68I0CQHL31jgy8ACGf6OlQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Overview of the statistical analysis. (A) For each amplicon i, we model the expected fraction of reads harboring the HbS allele in the total cfDNA isolated from the mother as a mixture of fetal (ψi) and maternal (ϕi) components in proportions determined by the fetal fraction w. The control shares the same expected fraction of mutated reads ϕi with the mother, which helps increase the precision of the estimates. (B) For each case, we estimate the expected fetal (ψ1,ψ2,ψ3) and maternal (ϕ1,(ϕ2,(ϕ3) fractions of mutated reads per amplicon. (C) We also estimate the overall distribution (given the data) of each triplet of expected maternal and fetal fractions presented in panel B. For a carrier mother (AS), the distribution of the maternal fractions is close to 50%, while for a fetus with the disease (SS), the corresponding distribution is shifted to the right of the maternal distribution. The stronger this shift E[P], the more likely it is that the fetus has the disease. (D) Overview of model training. We identified an optimal E[P] threshold equal to 0.62. The variance of the various performance metrics was estimated using the bootstrap. (E) Overview of applying the calibrated model on both the training and test cohorts. (F-H) Overview of model performance with decreasing fetal fraction. A false negative arises at a fetal fraction of 0.5%, while false positives arise at fetal fractions <4%. Four samples (in green) with E[P] scores close to the threshold are also characterized by small fetal fractions (≤0.9%). Statistical analysis was conducted in R10 and Stan.11,12 Details are given in supplemental Methods. TNR, true negative result; TPR, true positive result.
Overview of the statistical analysis. (A) For each amplicon i, we model the expected fraction of reads harboring the HbS allele in the total cfDNA isolated from the mother as a mixture of fetal (ψi) and maternal (ϕi) components in proportions determined by the fetal fraction w. The control shares the same expected fraction of mutated reads ϕi with the mother, which helps increase the precision of the estimates. (B) For each case, we estimate the expected fetal (ψ1,ψ2,ψ3) and maternal (ϕ1,(ϕ2,(ϕ3) fractions of mutated reads per amplicon. (C) We also estimate the overall distribution (given the data) of each triplet of expected maternal and fetal fractions presented in panel B. For a carrier mother (AS), the distribution of the maternal fractions is close to 50%, while for a fetus with the disease (SS), the corresponding distribution is shifted to the right of the maternal distribution. The stronger this shift E[P], the more likely it is that the fetus has the disease. (D) Overview of model training. We identified an optimal E[P] threshold equal to 0.62. The variance of the various performance metrics was estimated using the bootstrap. (E) Overview of applying the calibrated model on both the training and test cohorts. (F-H) Overview of model performance with decreasing fetal fraction. A false negative arises at a fetal fraction of 0.5%, while false positives arise at fetal fractions <4%. Four samples (in green) with E[P] scores close to the threshold are also characterized by small fetal fractions (≤0.9%). Statistical analysis was conducted in R10 and Stan.11,12 Details are given in supplemental Methods. TNR, true negative result; TPR, true positive result.
Our approach estimates the maternal and fetal fractions of reads harboring the HbS allele (Figure 2A-B). For a carrier mother (AS), this fraction is close to 50%, while for a fetus with the disease (SS), the corresponding fraction is shifted to the right (Figure 2C). The expected magnitude of this shift, E[P], ranges between 0 and 1, and it is used to predict the fetus as SS (homozygote), if sufficiently high. An optimal threshold for E[P] was determined by recruiting 29 subjects with known fetal disease status from IPT, followed by applying NIPT and calculating an E[P] value for each. Then, for E[P] threshold values between 0 and 1, we predicted the fetal disease status in each case and calculated the sensitivity (true positive rate), specificity (true negative rate), and Matthews correlation coefficient (MCC), a balanced metric for measuring the performance of binary classifiers (Figure 2D). We estimated the variance of all 3 metrics using the bootstrap (supplemental Methods). Increasing the E[P] threshold had opposite effects on sensitivity and specificity, reaching an optimal value of 62% at which MCC was maximized (Figure 2D). A second MCC maximum at 77% associated with lower sensitivity (higher false negative rate) was ignored. At the optimal E[P] threshold, our NIPT correctly identified 8 true positives, 17 true negatives, 1 false negative, and 3 false positives (supplemental Table 1; supplemental Figure 1) achieving 89% sensitivity, 85% specificity, 73% positive predictive value (PPV), and 94% negative predictive value (NPV).
In order to assess the predictive capacity of our NIPT, we recruited 28 additional subjects, who had also undergone IPT, and we applied our method on each using the previously determined optimal E[P] threshold of 62%. The test returned 7 true positives, 19 true negatives, 2 false positives, and no false negatives, achieving 100% sensitivity and NPV, 91% specificity, and 78% PPV (supplemental Table 1; supplemental Figure 1).
On all 57 subjects, the proposed NIPT called 5 false positives and 1 false negative (Figure 2E) corresponding to 94% sensitivity, 88% specificity, 75% PPV, and 98% NPV. The single false negative can be attributed to the very low fetal fraction (0.5%) of the corresponding case (Figure 2F). Furthermore, 4 samples close to the E[P] threshold (green dots in Figure 2E-G) are also associated with very low (≤0.9%) fetal fractions (Figure 2G). Overall, the NIPT demonstrates 100% sensitivity and NPV for fetal fractions >0.5% and 100% specificity and PPV for fetal fractions ≥4% (Figure 2H).
Our methodology can be used as a noninvasive screening tool in any clinical scenario in which the exclusion of SCA or any other recessive disorder is important. However, given its dependence on fetal fraction, the presence of an affected baby has to be confirmed by IPT.
We therefore expect that our test will be implemented similarly to noninvasive aneuploidy diagnosis to confidently exclude an affected pregnancy, thereby considerably reducing the number of IPTs performed to confirmatory testing of positive results only.
Our test is superior to previous methodologies as it does not require the paternal genotype and has 100% sensitivity for fetal fractions >0.5%. Therefore, only 2 out of 57 samples would have been rejected from analysis (supplemental Table 1). We successfully diagnosed genotypes as early as 8 weeks of gestation (range, 8-17 weeks; data not shown). The specificity of our method is 100% when fetal fractions are ≥4%. More efficient initial enrichment of fetal DNA therefore represents one way to avoid IPT altogether.
Our method will particularly appeal to countries with high incidence of SCA and a demand for prenatal diagnosis (ie, Nigeria), where access to IPT is limited due to the high cost and relatively low numbers of trained obstetricians. It would therefore make NIPT available to a larger number of beneficiaries.
We believe that the future adoption of this NIPT will also have utility in other clinical scenarios:
Treatment options for children and young adults who develop early complications from SCA include bone marrow or cord blood transplantation. Several clinical trials using autologous gene editing of stem cells also reported successful outcomes.13,14 Knowledge of the fetal genotype before birth would allow advance preparation for umbilical cord blood stem cell sampling for future cellular therapy, without placing the fetus at unnecessary risk through invasive testing.
Importantly, our method does not require prior knowledge of the father’s genotype, which will allow its straightforward incorporation into routine antenatal care with a rapid turnaround time.
The test could easily be adapted for detection of dominant monogenic disorders, de novo mutations, and other autosomal recessive conditions.
The online version of this article contains a data supplement.
Authorship
Contribution: A.C. performed experiments and data analysis; D.V.V. designed and implemented in software the statistical model and performed the statistical analysis; M.P. provided samples and assisted with clinical interpretation; F.S. and B.C. provided samples; and S.H. and A.S. invented technology, designed experiments, and led the data analysis and technical and clinical validation.
Conflict-of-interest disclosure: The technology presented here is subject of a Patent Cooperation Treaty patent application, plus a copyright protection for the statistical method and the data set, and all 3 pieces of the technology are available to license. A.S. and S.H. are coinventors of the intellectual property. A.S. received honoraria from Janssen, Gilead, Roche, and AbbVie and an unrestricted educational grant from Janssen. The remaining authors declare no competing financial interests.
Correspondence: Anna Schuh, Department of Oncology, University of Oxford, John Radcliffe Hospital, Level 4, Headington, Oxford OX39DU, United Kingdom; e-mail: anna.schuh@oncology.ox.ac.uk.
