

Key Points
Stroke is more common in TTP survivors than in an age- and sex-matched control population.
Lower ADAMTS13 activity after recovery from TTP is associated with stroke.

Abstract
With timely and effective treatment, most patients with thrombotic thrombocytopenic purpura (TTP) survive the acute TTP episode. In addition to the risk of relapse, TTP survivors have higher all-cause mortality than the general population and increased rates of chronic morbidities, including hypertension, depression, and mild cognitive impairment. We conducted this retrospective-prospective cohort study to determine the incidence and prevalence of stroke after recovery from acute TTP and to test the hypothesis that lower ADAMTS13 activity after recovery from TTP is associated with an increased risk of stroke during remission. Of 170 consecutive patients treated for TTP at The Johns Hopkins Hospital from 1995 through 2018, 14 (8.2%) died during the index episode and 19 were observed for less than 1 month after recovery. Of the remaining 137 patients, 18 (13.1%) developed stroke unrelated to an acute TTP episode over a median observation period of 3.08 years, which is fivefold higher than the expected prevalence of 2.6% from an age- and sex-matched reference population (P = .002). ADAMTS13 activity during remission was measured in 52 patients and was >70% in 44.2%, 40% to 70% in 23.1%, 10% to 39% in 25%, and <10% in 7.7%. Stroke after recovery from acute TTP occurred in 0% (0 of 22) of patients with normal remission ADAMTS13 activity (>70%) and in 27.6% (8 of 29) of patients with low ADAMTS13 activity (≤70%; P = .007). In conclusion, stroke is common after recovery from TTP and is associated with reduced ADAMTS13 activity during remission.
Introduction
Thrombotic thrombocytopenic purpura (TTP) is an acute, life-threatening disorder characterized by microangiopathic hemolytic anemia, thrombocytopenia, and ischemic organ failure. Acquired TTP is caused by an autoantibody-mediated deficiency of ADAMTS13, a von Willebrand factor (VWF)–cleaving protease,1 which results in circulating ultralarge VWF multimers that cause platelet aggregation and systemic microvascular thrombi. Plasma exchange has reduced the mortality caused by acute TTP episodes from >90% to <20%,2,3 and recent advances in targeted therapies for TTP, such as rituximab and caplacizumab, are expected to further improve outcomes of acute TTP.4,5 However, long-term adverse outcomes after recovery from TTP have been less well studied.
Until recently, TTP survivors were expected to return to their previous level of health, except for the 30% to 50% risk of relapse.6 However, recent data from the Oklahoma TTP registry indicate that TTP survivors have worse survival than age- and sex-matched controls7 and higher rates of comorbidities such as hypertension, obesity, and lupus.7 TTP survivors also have high rates of neurocognitive impairment,8,9 and magnetic resonance imaging (MRI) revealed evidence of small- or large-vessel ischemia in 39% (9 of 23) of patients after recovery from TTP. Intriguingly, lower ADAMTS13 activity is associated with stroke in individuals without TTP.10 Animal studies also indicate a role of ADAMTS13 in development and progression of ischemic stroke.11,12
We conducted this cohort study to determine the proportion of TTP survivors affected by stroke after recovery from acute TTP and to test the hypothesis that lower ADAMTS13 activity after recovery from acute TTP is associated with an increased risk of stroke.
Methods
Patients
The study cohort included 170 consecutive patients with acquired TTP who were treated at The Johns Hopkins Hospital (JHH) from 1995 to 2018. Before 2014, patients were identified from plasma exchange records from the therapeutic apheresis service and blood bank, whereas from 2014 to 2018, patients were prospectively enrolled in the Johns Hopkins Thrombotic Microangiopathy Registry13 and provided informed consent for the collection of longitudinal clinical data and biological specimens. (The 41 patients enrolled in the registry were also included in the plasma exchange records.) The diagnosis of TTP was based on the presence of thrombocytopenia (platelet count, <100 × 109/L), microangiopathic hemolytic anemia (hemoglobin level, <10 g/dL, along with schistocytes on the peripheral blood smear), a clinical course consistent with TTP (response to plasma exchange therapy), and the absence of alternative thrombotic microangiopathies, such as atypical hemolytic uremic syndrome or transplant-associated microangiopathy were excluded from the study. After 2006 (when the ADAMTS13 fluorescence resonance energy transfer assay was first offered at JHH), documented ADAMTS13 activity <10% was added to the diagnostic criteria. Only ADAMTS13 activity drawn prior to initiating plasma exchange was considered. A subset of patients who had their initial episode before 2006 had TTP relapses during which ADAMTS13 deficiency was documented (21 of 54 patients entering the cohort before 2006 had relapses, of which 8 had ADAMTS13 testing showing activity <10%). For the analysis of incident stroke after recovery from acute TTP, we excluded patients who died during the index TTP episode and those who had less than 1 month of follow-up after recovery from TTP. Patients were observed from TTP diagnosis (or first clinical contact after TTP diagnosis for patients who were transferred to our center after being diagnosed with TTP at other hospitals) until date of death or last clinical contact. The institutional review board at The Johns Hopkins University approved this study.
Data management and study outcomes
We extracted data from the electronic medical record, including patient demographics, details of TTP presentation, diagnosis and treatment, and the presence of comorbidities, including hypertension, diabetes mellitus, obesity (defined as body mass index >30 kg/m2), hyperlipidemia, atrial fibrillation, or chronic kidney disease (defined as a glomerular filtration rate <60 mL/min per 1.73 m2 persisting over at least 3 months),14 systemic lupus erythematosus (SLE), and other autoimmune diseases. In addition to laboratory studies during the acute episode, we recorded ADAMTS13 activity during remission, when available. Our current practice is to evaluate ADAMTS13 activity every 3 months, whenever possible. Only results of ADAMTS13 activity assays performed during clinical remission (with a normal platelet count and no clinical evidence of TTP) at least 3 months from the acute TTP episode were included in analyses of remission ADAMTS13 activity. All ADAMTS13 activity assays were performed at the Johns Hopkins Hospital Special Coagulation Laboratory using the fluorescence resonance energy transfer method with a standardized assay that has remained unchanged since it was first offered.15 The entire medical record including clinical notes from hospitalizations and outpatient visits, laboratory studies, and imaging studies was reviewed for each patient.
Stroke during clinical remission after recovery from TTP was recorded as the primary study outcome. A normal platelet count was used as a marker of TTP in remission; however, normal platelet count at presentation alone may not rule out recurrent TTP in all cases. To account for this, the clinical course after presenting with neurologic symptoms (and in the days before presenting, if available) was reviewed, and if consistent with a TTP recurrence (subsequent thrombocytopenia and evidence of microangiopathic hemolysis), the episode was excluded from the study end point of stroke during remission. Stroke was defined as a documented new neurologic deficit(s) with a corresponding ischemic lesion(s) on MRI of the brain. The determination of stroke was restricted to patients who presented with focal neurologic symptoms lasting for >24 hours (rather than also including transient focal symptoms), consistent with the World Health Organization definition of stroke.16 We added the requirement of a brain MRI showing a corresponding acute ischemic lesion (increased diffusion-weighted imaging signal or increased signal on T2-weighted image) to exclude an older stroke that may have occurred during or before acute TTP episodes. The determination of the study outcome (stroke) was initially made by 1 of 3 observers (H.U., J.K., and K.D.) who performed primary data abstraction. All outcomes were independently verified by a second observer who was blinded to the initial determination (S.C.). Separate from the study outcome, we recorded the presence of a stroke during acute TTP (as a possible association of subsequent stroke during remission) and included only overt neurologic symptoms lasting for >24 hours (rather than transient focal symptoms during a TTP episode) to identify these events. For patients who died, causes of death were recorded from the medical record.
Statistical analysis
Data were summarized as counts and percentages, medians, and interquartile range (IQR, representing the 25th-75th percentiles [Q25%-75%]). The observed prevalence of stroke (unrelated to an acute TTP episode) in TTP survivors was compared with expected rates determined on the basis of age- and sex-specific rates from a reference population, the National Health and Nutrition Examination Survey (NHANES) 2013-2016, using indirect standardization methods.17,18 In the TTP cohort, we tested differences by stroke status with a χ2 test for categorical covariates, and we examined continuous covariates with the Student t test or the Wilcoxon Mann-Whitney U test for normal and nonnormal distributions, respectively. We used Cox regression to evaluate risk factors for stroke including age, sex, and the presence of hypertension. Date of TTP diagnosis was set as time 0, and covariates were selected on the basis of known or suspected association with the outcome and results of univariate analysis. The proportional hazards assumption was assessed, and Martingale and deviance residuals were used to assess model assumptions and the effect of outlier cases.
To evaluate the association of remission ADAMTS13 activity with stroke, we used the average of remission ADAMTS13 activity measurements, defined as ADAMTS13 activity measured at least 3 months after an acute episode during clinical remission determined on the basis of a normal platelet count. We used the mean ADAMTS13 activity rather than the measurement closest to the stroke, because there would be no clear value of use for the majority of patients who did not develop stroke. Additionally, we used the mean rather than the median because it is likely to be a better measure of exposure to low ADAMTS13. We categorized mean remission ADAMTS13 activity as normal (>70%) and low (≤70%). This cutoff is based on the reference range at our institutional laboratory (70%-150%), which is based on healthy individuals. The cutoff also correlates with the median of the lowest quartile ADAMTS13 activity (71%), which was associated with an increased risk of ischemic stroke in the population-based Rotterdam cohort.10 We used exact logistic regression to evaluate the association of remission ADAMTS13 activity with stroke adjusted for age, sex, and comorbidities such as hypertension and diabetes. We chose exact logistic regression over logistic regression because there were no stroke events in the remission ADAMTS13 >70% category and logistic regression, which relies on maximum likelihood estimation, does not provide a finite regression coefficient. Exact regression is an alternative method that can provide finite, consistent estimates of regression parameters (median unbiased estimates) when maximum likelihood estimates cannot be computed because of complete separation.19 We also analyzed the data as a nested case-control study using incidence-density sampling to account for time since TTP diagnosis. P < .05 was considered significant for all analyses. All analyses were performed with STATA version 16 (STATA Corp, College Station, TX).
Results
Cohort demographics
Between 1995 and 2018, 170 patients with a confirmed diagnosis of TTP were treated for 248 episodes of TTP at JHH. Seventy-two (52.5%) patients entered the cohort at their first TTP episode, whereas 65 (47%) had had an episode before being seen at JHH. Fourteen patients (8.2%) died during the first episode of TTP treated at JHH. Of the 156 survivors, 137 were observed for at least 1 month after recovery from TTP and were included in the analysis (Figure 1). Median follow-up was 3.08 (IQR, 0.66, 7.79) years with a total of 700.7 patient-years of observation. Median age at the end follow-up was 48.8 (IQR, 35.3, 60.3) years. Sixty-two percent of the cohort was African American and 67.9% were female.
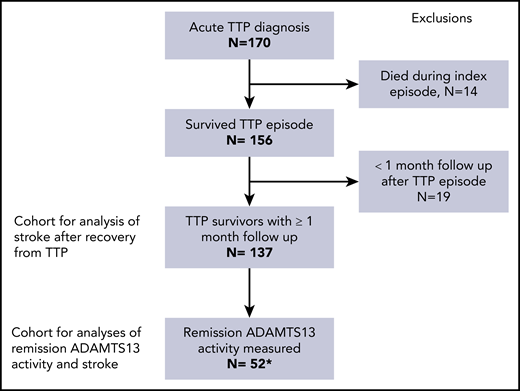
Flowchart of individuals included in the analysis. Between 1995 and 2018, 170 individual patients were treated for a confirmed diagnosis of TTP. Of these, 14 died during the index TTP episode, and another 19 had less than 1 month of follow-up after recovery from TTP. The remaining 137 patients were included in the analyses of stroke after TTP. A subset of 52 patients had available measurements of ADAMTS13 activity in remission, and could be evaluated for association of remission ADAMTS13 with subsequent stroke. *One of the 52 patients had had a stroke before the first measurement of remission ADAMTS13 and was excluded from this analysis.
Flowchart of individuals included in the analysis. Between 1995 and 2018, 170 individual patients were treated for a confirmed diagnosis of TTP. Of these, 14 died during the index TTP episode, and another 19 had less than 1 month of follow-up after recovery from TTP. The remaining 137 patients were included in the analyses of stroke after TTP. A subset of 52 patients had available measurements of ADAMTS13 activity in remission, and could be evaluated for association of remission ADAMTS13 with subsequent stroke. *One of the 52 patients had had a stroke before the first measurement of remission ADAMTS13 and was excluded from this analysis.
TTP patients have higher stroke prevalence than age- and sex-matched controls
Stroke during follow-up after recovery from TTP, unrelated to an acute TTP episode, occurred in 13.1% (18 of 137) of individuals, which is higher than the expected prevalence of 2.6%, based on age- and sex-stratified estimates from NHANES 2013-2016 (P = .002; Figure 2). Incidence of stroke in our cohort was 2.57 per 100 patient years. Median time from first TTP diagnosis to stroke for these patients was 2.8 (IQR, 0.8, 10.0) years.
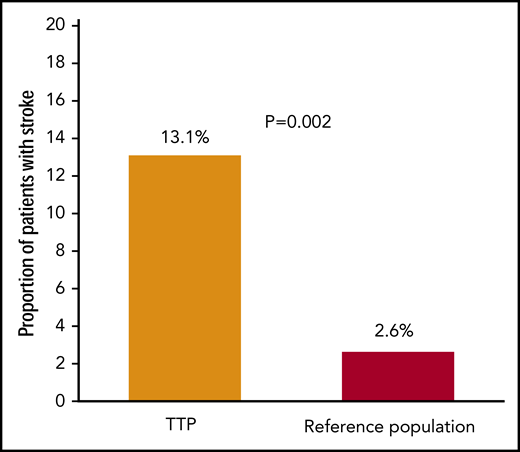
Proportion of TTP survivors who developed stroke during remission. The proportion of TTP survivors affected by stroke (unrelated to an acute TTP episode) was significantly higher than expected based on an age- and sex-matched control reference population (United States, NHANES 2013-2016).
Proportion of TTP survivors who developed stroke during remission. The proportion of TTP survivors affected by stroke (unrelated to an acute TTP episode) was significantly higher than expected based on an age- and sex-matched control reference population (United States, NHANES 2013-2016).
On univariate analysis, TTP patients who developed stroke were older than those without stroke (median age, 57.2 vs 46.7 years; P = .021) and had a higher proportion of comorbid hypertension (72.2% vs 41.2%; P = .021). However, the proportion of patients with stroke during acute TTP, as well as other comorbidities, including diabetes mellitus, obesity, dyslipidemia, atrial fibrillation, chronic kidney disease, lupus, and other autoimmune disorders was not significantly different in the stroke group (Table 1). Patients who developed stroke during remission from TTP were also more likely to have had stroke during the acute TTP episode than were patients without stroke during TTP remission, though the difference did not reach statistical significance (16.7% vs 8.4%; P = .379). In a multivariate Cox regression model, age (hazard ratio [HR], 1.01 [95% confidence interval (CI), 0.97-1.04]; P = .772), male sex (HR, 1.08 [95% CI, 0.29-3.98]; P = .901), hypertension (HR, 1.21 [95% CI, 0.39-3.71]; P = .735), and stroke during the acute TTP episode (HR, 1.28 [95% CI, 0.31-5.21]; P = .733) were not associated with incident stroke; however, this finding may be attributable, at least in part, to the small sample size and small number of events. Table 2 summarizes characteristics of stroke episodes in 18 affected patients.
Demographics and comorbidities of patients with and without stroke
| Characteristic | Total (N = 137) | No stroke (n = 119) | Stroke (n = 18) | P* |
|---|---|---|---|---|
| Age, median (IQR), y | 48.8 (35.3, 60.3) | 46.7 (34.9, 58.9) | 57.2 (49.9, 67.3) | .028 |
| Race, % | ||||
| White | 32.8 | 33.6 | 27.8 | .685 |
| African American | 62.0 | 60.5 | 72.2 | |
| Other | 5.0 | 5.8 | 0 | |
| Female sex, % | 67.9 | 65.5 | 83.3 | .178 |
| Stroke during acute TTP episode, % | 9.5 | 8.4 | 16.7 | .379 |
| Total number of TTP episodes, median, IQR | 1 (1, 2) | 1 (1, 2) | 2 (1, 2) | .083 |
| Hypertension, % | 45.3 | 41.2 | 72.2 | .021 |
| Diabetes mellitus, % | 22.6 | 20.2 | 38.9 | .126 |
| Obesity (BMI >30), % | 29.2 | 30.3 | 22.2 | .347 |
| Dyslipidemia, % | 17.5 | 15.1 | 33.3 | .090 |
| Atrial fibrillation, % | 5.8 | 5.9 | 5.6 | .956 |
| SLE, % | 8.0 | 6.7 | 16.7 | .160 |
| Other autoimmune disorder, % | 8.0 | 6.7 | 16.7 | .160 |
| Chronic kidney disease, % | 16.8 | 16.0 | 22.2 | .737 |
| Current smoking, % | 14.6 | 12.7 | 22.2 | .220 |
| Characteristic | Total (N = 137) | No stroke (n = 119) | Stroke (n = 18) | P* |
|---|---|---|---|---|
| Age, median (IQR), y | 48.8 (35.3, 60.3) | 46.7 (34.9, 58.9) | 57.2 (49.9, 67.3) | .028 |
| Race, % | ||||
| White | 32.8 | 33.6 | 27.8 | .685 |
| African American | 62.0 | 60.5 | 72.2 | |
| Other | 5.0 | 5.8 | 0 | |
| Female sex, % | 67.9 | 65.5 | 83.3 | .178 |
| Stroke during acute TTP episode, % | 9.5 | 8.4 | 16.7 | .379 |
| Total number of TTP episodes, median, IQR | 1 (1, 2) | 1 (1, 2) | 2 (1, 2) | .083 |
| Hypertension, % | 45.3 | 41.2 | 72.2 | .021 |
| Diabetes mellitus, % | 22.6 | 20.2 | 38.9 | .126 |
| Obesity (BMI >30), % | 29.2 | 30.3 | 22.2 | .347 |
| Dyslipidemia, % | 17.5 | 15.1 | 33.3 | .090 |
| Atrial fibrillation, % | 5.8 | 5.9 | 5.6 | .956 |
| SLE, % | 8.0 | 6.7 | 16.7 | .160 |
| Other autoimmune disorder, % | 8.0 | 6.7 | 16.7 | .160 |
| Chronic kidney disease, % | 16.8 | 16.0 | 22.2 | .737 |
| Current smoking, % | 14.6 | 12.7 | 22.2 | .220 |
BMI, body mass index.
Fisher's exact test (categorical variables), Mann-Whitney U test (continuous variables).
Details of stroke during remission after recovery from TTP (n = 18)
| Patient no. | Sex/age at stroke, y | Time from TTP to stroke, mo | Clinical presentation and MRI/MRA findings | Stroke during acute TTP | Platelet count at stroke diagnosis, ×109/L | LDH at stroke | Remission ADAMTS13 activity near stroke* |
|---|---|---|---|---|---|---|---|
| 1 | F/43 | 7 | Gait abnormalities, imbalance, and cerebellar stroke on MRI/MRA. | No | 156 | 350† | 63 |
| 2 | M/23 | 2 | Right hemiparesis, aphasia, and left MCA stroke on MRI/MRA. | Yes | 186 | N/A | 51 (+1 mo) |
| 3 | M/55 | 8 | Right hemiparesis and expressive aphasia; large left MCA infarct on MRI/MRA. | No | 276 | N/A | 45 (+4 mo) |
| 4 | F/52 | 87 | Left hemiparesis, posterior MCA infarct on MRI/MRA. | No | 179 | 244 | 29 (+3 mo) |
| 5 | F/31 | 12 | Aphasia, right hemiparesis, and left MCA infarct on MRI/MRA. | No | 213 | N/A | N/A |
| 6 | F/49 | 45 | Left upper extremity weakness, right precentral gyrus infarct on MRI; no MRA lesion identified. | No | 201 | 150 | 30 |
| 7 | F/51 | 140 | Diplopia, facial numbness, infarcts in centrum semiovale and pons on MRI, no MRA abnormalities. | No | 249 | N/A | N/A |
| 8 | F/26 | 1 | Aphasia, and left MCA infarct on MRI/MRA. | No | 176 | N/A | 59 |
| 9 | F/20 | 66 | Left arm weakness; right MCA infarct on MRI/MRA. | No | 283 | N/A | N/A |
| 10 | F/41 | 19 | Right hemiparesis, seizure, left MCA infarct on MRI. | No | 177 | 249 | 42 (−1 wk) |
| 11‡ | F/64 | 41 | Aphasia. MRA showed acute infarcts of the left posterior perisylvian, parieto-occipital, and parietal lobes in addition to chronic microvascular disease. MRA was suboptimal due to patient motion but showed irregular narrowing of both MCA with prominent loss of flow in the distal branches of the left MCA. | No | 150 | 277 | N/A |
| 12 | F/67 | 59 | Right hemiparesis followed by obtundation. Died due to massive left MCA stroke, with hemorrhagic conversion on MRI. | No | 217 | 248 | N/A |
| 13‡ | F/75 | 173 | Right upper extremity weakness, aphasia, and left MCA and ACA stroke. | No | 302 | 199 | N/A |
| 14 | F/72 | 165 | Right hemiparesis; left MCA stroke. | No | 180 | 237 | N/A |
| 15‡ | F/49 | 22 | Left leg weakness, dysarthria, and dysphagia. Acute pontine stroke on MRI, no lesions on MRA. | No | 188 | 211 | 25 |
| 16‡ | F/60 | 36 | Slurred speech, facial droop, blurry vision, leg weakness, and unstable gait; biparietal and left cerebellar acute infarcts on MRI. | Yes | 164 | 173 | N/A |
| 17 | F/59 | 120 | Aphasia, right hemiparesis, left MCA infarct on MRI/MRA. | No | 265 | N/A | N/A |
| 18 | F/50 | 17 | Expressive aphasia, right hand weakness; acute temporoparietal infarction; MRA showed no abnormality in circle of Willis and carotid arteries. | Yes | 159 | 231 | N/A |
| Patient no. | Sex/age at stroke, y | Time from TTP to stroke, mo | Clinical presentation and MRI/MRA findings | Stroke during acute TTP | Platelet count at stroke diagnosis, ×109/L | LDH at stroke | Remission ADAMTS13 activity near stroke* |
|---|---|---|---|---|---|---|---|
| 1 | F/43 | 7 | Gait abnormalities, imbalance, and cerebellar stroke on MRI/MRA. | No | 156 | 350† | 63 |
| 2 | M/23 | 2 | Right hemiparesis, aphasia, and left MCA stroke on MRI/MRA. | Yes | 186 | N/A | 51 (+1 mo) |
| 3 | M/55 | 8 | Right hemiparesis and expressive aphasia; large left MCA infarct on MRI/MRA. | No | 276 | N/A | 45 (+4 mo) |
| 4 | F/52 | 87 | Left hemiparesis, posterior MCA infarct on MRI/MRA. | No | 179 | 244 | 29 (+3 mo) |
| 5 | F/31 | 12 | Aphasia, right hemiparesis, and left MCA infarct on MRI/MRA. | No | 213 | N/A | N/A |
| 6 | F/49 | 45 | Left upper extremity weakness, right precentral gyrus infarct on MRI; no MRA lesion identified. | No | 201 | 150 | 30 |
| 7 | F/51 | 140 | Diplopia, facial numbness, infarcts in centrum semiovale and pons on MRI, no MRA abnormalities. | No | 249 | N/A | N/A |
| 8 | F/26 | 1 | Aphasia, and left MCA infarct on MRI/MRA. | No | 176 | N/A | 59 |
| 9 | F/20 | 66 | Left arm weakness; right MCA infarct on MRI/MRA. | No | 283 | N/A | N/A |
| 10 | F/41 | 19 | Right hemiparesis, seizure, left MCA infarct on MRI. | No | 177 | 249 | 42 (−1 wk) |
| 11‡ | F/64 | 41 | Aphasia. MRA showed acute infarcts of the left posterior perisylvian, parieto-occipital, and parietal lobes in addition to chronic microvascular disease. MRA was suboptimal due to patient motion but showed irregular narrowing of both MCA with prominent loss of flow in the distal branches of the left MCA. | No | 150 | 277 | N/A |
| 12 | F/67 | 59 | Right hemiparesis followed by obtundation. Died due to massive left MCA stroke, with hemorrhagic conversion on MRI. | No | 217 | 248 | N/A |
| 13‡ | F/75 | 173 | Right upper extremity weakness, aphasia, and left MCA and ACA stroke. | No | 302 | 199 | N/A |
| 14 | F/72 | 165 | Right hemiparesis; left MCA stroke. | No | 180 | 237 | N/A |
| 15‡ | F/49 | 22 | Left leg weakness, dysarthria, and dysphagia. Acute pontine stroke on MRI, no lesions on MRA. | No | 188 | 211 | 25 |
| 16‡ | F/60 | 36 | Slurred speech, facial droop, blurry vision, leg weakness, and unstable gait; biparietal and left cerebellar acute infarcts on MRI. | Yes | 164 | 173 | N/A |
| 17 | F/59 | 120 | Aphasia, right hemiparesis, left MCA infarct on MRI/MRA. | No | 265 | N/A | N/A |
| 18 | F/50 | 17 | Expressive aphasia, right hand weakness; acute temporoparietal infarction; MRA showed no abnormality in circle of Willis and carotid arteries. | Yes | 159 | 231 | N/A |
ACA, anterior cerebral artery; F, female; M, male; LDH, lactate dehydrogenase; MCA, middle cerebral artery; MRA, magnetic resonance angiography; N/A, not available.
Only remission ADAMTS13 activity within 3 months of the stroke is included in this table. If there are no times in parenthesis, then the ADAMTS13 activity was checked at stroke presentation. The time in parenthesis indicates the interval from stroke to ADAMTS13 activity evaluation.
Patient 1 had persistently elevated LDH levels at stroke diagnosis but did not develop recurrent TTP. The LDH elevation was attributed to severe autoimmune myositis in the setting of SLE (peak creatine kinase 1940 U/L and peak LDH 770 U/L), which resolved after treatment with corticosteroids and methotrexate. ADAMTS13 activity was 63%, which also argues against ongoing TTP.
Patients who had recurrent strokes after recovery from TTP.
Low ADAMTS13 activity in remission is common and is associated with stroke
ADAMTS13 activity during clinical remission at least 3 months after an acute TTP episode was measured in 52 patients. Characteristics of patients included and excluded from the remission ADAMTS13 analysis are shown in supplemental Table S1, available on the Blood Web site. The median number of ADAMTS13 activity measurements during remission was 3 (range, 1-18). Remission ADAMTS13 activity was normal (>70%) in 44.2% (23 of 52), 40% to 70% in 23.1% (12 of 52), 10% to 39% in 25% (13 of 52), and <10% in 7.7% (4 of 52). One patient did not have ADAMTS13 measured until 5 years after her stroke (which occurred the month following initial TTP diagnosis). Since the outcome (stroke) occurred before measurement of the exposure (remission ADAMTS13 activity), this patient was excluded from the analysis of ADAMTS13 in remission as a risk factor for subsequent stroke. Among the 51 evaluable patients, stroke after recovery from acute TTP occurred in 0% (0 of 22) of patients with normal mean ADAMTS13 activity (>70%) during remission and in 27.6% (8 of 29) of patients with low ADAMTS13 activity (≤70%) during remission (P = .007). Figure 3 shows the distribution of mean remission ADAMTS13 activity in patients with and without stroke during TTP remission. When we further categorized ADAMTS13 activity, stroke occurred in 0% (0 of 4) of patients with remission ADAMTS13 <10%; in 38.5% (5 of 13) with remission ADAMTS13 activity 10% to 39%; in 25.0% (3 of 12) with remission ADAMTS13 activity 40% to 70%; and in 0% (0 of 22) with remission ADAMTS13 activity >70%. Using exact logistic regression, remission ADAMTS13 activity was significantly associated with stroke after recovery from TTP (odds ratio [OR], 11.06 [95% CI, 1.31 to +infinity]; P = .024) after adjusting for sex (OR, 2.52 [95% CI, 0.20-50.04]; P = .741), age (OR, 0.97 [95% CI, 0.89-1.05]; P = .559), hypertension (OR, 12.99 [95% CI, 1.34 to +infinity]; P = .025), and number of TTP episodes (OR, 4.14 [95% CI, 0.95-44.20]; P = .067). Since the median duration of follow-up was slightly higher in cases (4.43 [IQR, 1.00, 11.79] years) than in controls (3.91 [IQR, 1.16, 7.75] years), we also analyzed the 51 evaluable patients as a nested case-control study using incidence-density sampling, which also supported the association of mean remission ADAMTS13 activity with stroke (supplemental data).
![Average remission ADAMTS13 activity in patients without stroke (n = 43) and with stroke (n = 8). Median average remission ADAMTS13 activity was lower in patients with stroke vs those who did not develop stroke during follow-up (33% [IQR, 20%, 49%] vs 71% [IQR, 33%, 96%]; P = .021). The dashed line represents ADAMTS13 activity of 70%, where an arbitrary threshold of >70% is considered normal.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/134/13/10.1182_blood.2019001056/3/m_bloodbld2019001056f3.png?Expires=1767703266&Signature=AhyOmWUP8s0SVEXhXkNT3shmonPCt1ZKasZZUZfO-W2P5MD4T83lvd4Mcrf5H-BzKVtJLme-27UpyYKShtnKFIJoJg8x2ztbuZjMSJrjskcBeW5dOeH9QywiVmJZq0cxJPvzfe1Kee271aMqw8MhOXdxGgqbklmm4QoiiFCOcWuHg1-ZXjkhsinrclPRURGhKSVYXKM7cmJbLufXL8pwh9z~EilJrpNSCa2ehl~hqPG5vGQO6uZp0r2ogrh8M28USNVYaUZwj0DgPutqXQXXR9SHc-UsrtYaPGF8Oi6Moskmb2QRHA96YeaOai6V~2yOT~u1oRlYvrZI9fdgb-fKdw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Average remission ADAMTS13 activity in patients without stroke (n = 43) and with stroke (n = 8). Median average remission ADAMTS13 activity was lower in patients with stroke vs those who did not develop stroke during follow-up (33% [IQR, 20%, 49%] vs 71% [IQR, 33%, 96%]; P = .021). The dashed line represents ADAMTS13 activity of 70%, where an arbitrary threshold of >70% is considered normal.
Average remission ADAMTS13 activity in patients without stroke (n = 43) and with stroke (n = 8). Median average remission ADAMTS13 activity was lower in patients with stroke vs those who did not develop stroke during follow-up (33% [IQR, 20%, 49%] vs 71% [IQR, 33%, 96%]; P = .021). The dashed line represents ADAMTS13 activity of 70%, where an arbitrary threshold of >70% is considered normal.
Mortality and causes of death
Over the follow-up period, 10.2% (14 of 137) patients died. Causes of death included TTP relapse (n = 2, 14.2%), ischemic stroke (n = 2, 14.2%), myocardial infarction (n = 2, 14.2%), malignancy (n = 5, 35.7%), and other causes (n = 3; 1 each with hypoxic respiratory failure, fungal pneumonia in the setting of HIV, or sepsis with HIV infection).
Discussion
With effective treatments, most patients with acquired TTP now survive an acute TTP episode. This has led to increasing scrutiny of long-term outcomes in this population.20 Increased rates of depression,7,21 cognitive impairment,8,22 hypertension TTP,7,23 poor quality of life,9 and reduced survival7 are reported in TTP survivors. In our cohort of 137 patients observed after recovery from TTP, the proportion of patients with stroke (unrelated to TTP recurrences) was significantly higher than expected, based on a reference US population. We restricted our analyses to stroke events that occurred outside of acute TTP episodes. When stroke occurring during TTP was included, overall stroke prevalence in this population was even higher (∼20%). Reduced ADAMTS13 activity during clinical remission after recovery from TTP was associated with subsequent stroke.
Less than half of the patients in our cohort had consistently normal ADAMTS13 activity during remission, a number that is concordant with results from the Oklahoma TTP registry.7 The association of lower ADAMTS13 activity in remission with stroke is particularly interesting because ADAMTS13 activity in remission is a potentially modifiable risk factor (for example, with rituximab) and because it may identify patients at high risk for stroke who may benefit from measures such as antiplatelet therapy and closer monitoring. Data from the Hereditary TTP Registry also lend credence to a significant role for ADAMTS13 in arterial thromboembolism; 28% of patients with congenital TTP had suffered an arterial thromboembolic event by the time of enrollment, including 21% with stroke.24
The association between ADAMTS13 and ischemic stroke was first described in small case-control studies of patients without TTP,25,26 and the Rotterdam Stroke Study showed an association between lower ADAMTS13 activity and stroke in a large population-based cohort of nearly 6000 older adults.10 Reduced ADAMTS13 activity has been shown to increase risk for cardiovascular disease in general,26,27 as well as all-cause and cardiovascular mortality.28 Studies in mice also support the role of ADAMTS13 in development and progression of ischemic stroke.11,12,29,-31 The most likely explanation for the risk of stroke associated with low ADAMTS13 is an accumulation of larger, more procoagulant VWF multimers,25,32 which leads to increased platelet activation and aggregation,33 complement activation on vascular endothelium,34,-36 and possible acceleration of atherosclerosis.37,-39 Studies in the general population have already identified VWF levels as a risk factor for stroke.25,32 We suggest that remission ADAMTS13 activity at levels of 10% to 70% may contribute to a state of subclinical platelet and vascular activation and cumulative vascular damage that leads to earlier arterial thrombotic events (Figure 4).
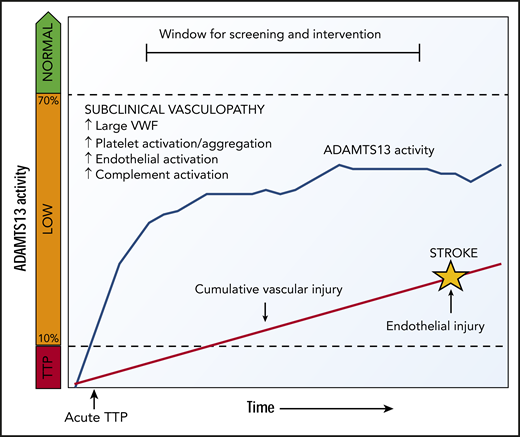
Proposed model for low ADAMTS13 activity causing vascular complications. After recovery from acute TTP, ADAMTS13 activity that increases but not does not reach normal levels may cause VWF-mediated platelet and vascular activation and cumulative vascular injury that leads to premature arterial thrombotic events such as stroke.
Proposed model for low ADAMTS13 activity causing vascular complications. After recovery from acute TTP, ADAMTS13 activity that increases but not does not reach normal levels may cause VWF-mediated platelet and vascular activation and cumulative vascular injury that leads to premature arterial thrombotic events such as stroke.
Another explanation of the high rate of stroke in our TTP cohort is that TTP patients have a relatively higher frequency of African American race, hypertension, and lupus, all of which are risk factors for stroke in the general population. In our cohort, hypertension was associated with stroke. Patients who developed stroke had higher rates of lupus and smoking and a slightly higher number of lifetime TTP episodes; however, these differences did not achieve statistical significance, most likely because of the small sample size. ADAMTS13 activity and traditional cardiovascular risk factors are not mutually exclusive and may work additively to increase the risk of ischemic cerebrovascular events.
Recovery of ADAMTS13 activity after an acute episode of immune TTP depends on suppression of anti-ADAMTS13 antibodies. However, the timeline of recovery of ADAMTS13 activity after an episode of TTP is poorly understood. Rituximab is increasingly used for the treatment of acute TTP and is likely to accelerate ADAMTS13 recovery after acute TTP.40,41 However, it is not known whether achieving normal ADAMTS13 activity quickly after an acute TTP episode translates into maintaining normal ADAMTS13 activity over longer periods (such as 1, 5, or 10 years). There is increasing recognition that low ADAMTS13 activity in remission is a risk factor for relapse,42 and preemptive rituximab may be used to prevent relapses in patients with severe ADAMTS13 deficiency (activity <10%) during remission.43 An intriguing area of further research involves evaluation of stroke rates in cohorts with higher rates of upfront or preemptive treatment with rituximab. TTP survivors also exhibit cognitive impairment,8,22 which may be caused by subclinical (silent cerebral infarcts) or overt stroke.44
Our study is limited by the relatively small size of our cohort, a common issue for single-center rare-disease cohorts, which prevented extensive evaluation of risk factors for stroke. The median follow-up of our cohort was also relatively short at ∼3 years, and it is likely that the lifetime burden of stroke in our cohort will continue to accrue. Additionally, we included only stroke for which patients were hospitalized at JHH, which likely underestimated the true proportion of patients who develop stroke after recovery from TTP. We were unable to establish the natural history of ADAMTS13 activity after recovery from acute TTP, because the frequency of measurement varied. ADAMTS13 activity was not routinely measured at the time of stroke diagnosis, so we cannot determine whether it is cumulative exposure to low ADAMTS13 activity, magnitude of ADAMTS13 deficiency, or both, that increase stroke risk.
In summary, TTP survivors have a high rate of stroke, even after recovery from their acute TTP episode, which is independent of TTP recurrence. Incomplete recovery of ADAMTS13 after an acute episode of TTP is associated with an increased risk of stroke and may provide an opportunity for screening and early intervention. It is possible that neurologic injury can be detected earlier, for example, as silent cerebral infarcts, ischemic lesions on MRI in the absence of corresponding neurologic deficits. These are associated with the risk of subsequent overt stroke in the general population44 and in patients with other hematologic disorders such as sickle cell disease.45 Optimal screening strategies for TTP patients at risk for cerebrovascular events are not established, but carotid Doppler ultrasonography, brain MRI, traditional cardiovascular disease risk factors,46 or a combination of these may be useful. Depending on the etiology of cerebrovascular events after TTP, the role of antiplatelet therapy or even ADAMTS13-directed therapy for primary and secondary prevention could be investigated; however, these treatments must be studied in randomized trials. TTP is more than an acute illness, and our results highlight the need for continued monitoring after recovery to manage long-term sequelae of TTP and the need to develop strategies for prevention and treatment of the neurologic sequelae of TTP.
Data sharing policy: Deidentified data can be made through an e-mail to the corresponding author/principal investigator (PI) (S.C.). Data Deidentification: The study team at Johns Hopkins will be the official repository of all study data. Any identifying data (Health Insurance Portability and Accountability Act–designated 17 protected health information elements) will be deleted prior to the release of any data to an internal or external investigator. These items include: names, addresses, telephone numbers, medical record numbers, photographs, date of birth, dates of hospital admission and discharge, date of death, or any other information that may uniquely identify an individual. Publications from the study will not contain any information that could potentially identify any participant in the study. In general, there should be no reason to include any form of patient listings in the study. If there is a compelling reason to include a patient listing, none of the potentially identifying information listed above (eg, date of birth, date of hospital discharge, and other information) will be listed. Data File Format Standard: CSV data files will be generated. These data files will have data formats for categorical variables included in the file, as well as complete variable labels. REDCap is capable of generating a CSV data file that can then be opened and imported with SAS, SPSS, Excel, and R statistical programs. Requesting Data Files: Both internal and external investigators are required to submit a proposal/e-mail requesting a dataset from the study. The proposal must describe the hypothesis or investigation to be conducted with the dataset. The PI will review the proposal for feasibility and for priority. If the request is reasonable, the contact PI and lead statistician will send a confidentiality agreement stipulating that the data file is to be used only for the purpose described in the proposal and that the requesting investigator must retain control over the file at all times. The requesting investigator is further required to supply annual progress reports on the analysis/investigation and to supply draft manuscripts. Prior to the release of the file, the contact PI and statistician will personally review the file and certify the level of deidentification.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This study was supported by a Mentored Research Award from the Hemostasis and Thrombosis Research Society and Johns Hopkins University Clinician Scientist Award (S.C.).
Authorship
Contribution: H.U. collected data and wrote the first draft of the manuscript; J.K. and K.D. collected data; A.R.M. and R.A.B. interpreted the analysis; T.S.K. oversaw the validation of test methodology and test performance and critically reviewed the manuscript; S.S., C.J.S., and E.M.B. critically reviewed the manuscript; M.B.S. and R.F.G. critically reviewed and edited the manuscript; S.C. designed the research, verified all outcomes, performed and interpreted the analyses, and wrote portions of the manuscript; and all authors read and approved the final draft of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Shruti Chaturvedi, Division of Hematology, Department of Medicine, The Johns Hopkins University School of Medicine, 720 Rutland Ave, Ross Research Building, Room 1025, Baltimore, MD 21205; e-mail: schatur3@jhmi.edu.
