

Key Points
PD-1 blockade using pembrolizumab administered after ASCT has an acceptable safety profile.
This treatment results in a high PFS in patients with cHL, including in high-risk patients.

Abstract
Autologous stem cell transplantation (ASCT) remains the standard of care for patients with relapsed/refractory (RR) classical Hodgkin lymphoma (cHL) who respond to salvage chemotherapy. However, relapse after ASCT remains a frequent cause of treatment failure, with poor subsequent prognosis. Because cHL is uniquely vulnerable to programmed cell death-1 (PD-1) blockade, PD-1 blockade given as consolidation after ASCT could improve ASCT outcomes. We therefore conducted a multicohort phase 2 study of pembrolizumab in patients with RR cHL after ASCT, hypothesizing that it would improve the progression-free survival (PFS) at 18 months after ASCT (primary end point) from 60% to 80%. Pembrolizumab was administered at 200 mg IV every 3 weeks for up to 8 cycles, starting within 21 days of post-ASCT discharge. Thirty patients were treated on this study. The median age was 33 years, and 90% were high-risk by clinical criteria. Seventy-seven percent completed all 8 cycles. Toxicity was manageable, with 30% of patients experiencing at least 1 grade 3 or higher adverse event (AE), and 40% at least 1 grade 2 or higher immune-related AE. Two patients were lost to follow-up in complete remission at 12 months. The PFS at 18 months for the 28 evaluable patients was 82%, meeting the primary end point. The 18-month overall survival was 100%. In conclusion, pembrolizumab was successfully administered as post-ASCT consolidation in patients with RR cHL, and resulted in a promising PFS in a high-risk patient cohort, supporting the testing of this strategy in a randomized trial. This trial was registered at www.clinicaltrials.gov as #NCT02362997.
Introduction
Most patients with classical Hodgkin lymphoma (cHL) are cured with frontline multiagent chemotherapy. For those with relapsed/refractory (RR) disease, the current standard of care is salvage chemotherapy followed by autologous stem cell transplantation (ASCT) for patients whose disease remits with salvage.1 However, up to one-half of transplanted patients will still relapse, with generally poor outcomes.2 Brentuximab vedotin (BV), a toxin-conjugated anti-CD30 monoclonal antibody (mAb), improves the progression-free survival (PFS) in high-risk patients with RR cHL undergoing ASCT, but, even with BV consolidation, ∼40% of patients will experience treatment failure within 5 years.3 There is therefore an important need to improve the outcome of ASCT in this patient population.
Therapeutic blockade of the programmed cell death-1 (PD-1) axis is an important new advance in oncology and is specifically an effective therapy in RR cHL. The malignant Hodgkin Reed Sternberg (HRS) cells very frequently harbor genetic amplification at 9p24.1, leading to overexpression of the PD-1 ligands, PD-L1 and PD-L2, on the tumor cell surface.4,5 Additionally, there is increased PD-L1 expression in tumor-associated macrophages, likely due to local interferon γ–mediated induction.6 This likely underlies the unique vulnerability of cHL to anti–PD-1 mAbs, as demonstrated by several studies in patients with RR cHL who had relapsed after or were ineligible for ASCT, with objective response rates around 70%.7-10 Yet, despite this high activity, most patients treated in the RR setting will eventually experience treatment failure. It may therefore be fruitful to deploy PD-1 blockade in earlier phases of treatment in an attempt to increase cure rates. The post-ASCT setting is a state of minimal residual disease, with active immune remodeling. Although ASCT has traditionally been viewed as primarily a cytotoxic modality, there is increasing recognition of the immunogenic effect of chemotherapy, which may also apply to ASCT,11,12 suggesting that consolidation treatment with PD-1 blockade could potentially improve the PFS of patients undergoing ASCT for RR cHL. We therefore designed a phase 2 study of pembrolizumab, a humanized immunoglobulin G4 anti-PD-1 mAb, used as consolidation post-ASCT.
Methods
Patients and centers
This phase 2, investigator-initiated, open-label, multicohort, multicenter clinical trial enrolled patients at 6 centers in the United States. The study accrued patients in 3 cohorts: 1 for cHL, 1 for diffuse large B-cell lymphoma, and 1 for T-cell lymphoma. Here, we present the results of the cHL cohort. This cohort enrolled patients ≥18 years of age with cHL who had relapsed after or were refractory to frontline therapy. They had to have received ASCT and had chemosensitive disease, that is, they had to have achieved partial or complete metabolic response after salvage therapy and prior to ASCT. Availability of a postsalvage, pre-ASCT positron emission tomography (PET) scan was required. Patients could not have received >3 prior lines of therapy (not counting ASCT). No additional therapy (radiotherapy, immunotherapy, or chemotherapy) was allowed after ASCT prior to study enrollment. In addition, patients had to have an Eastern Cooperative Oncology Group (ECOG) performance status <2 and adequate hematologic and organ function. Patients with central nervous system involvement of lymphoma, active or history of autoimmune disease, and active or prior pneumonitis were excluded. Patients who received prior treatment with an anti–PD-1, anti–PD-L1, or anti–CTLA-4 agent were permitted to enter the trial so long as they entered clinical remission with 1 of these agents prior to ASCT without intervening relapse. All patients signed informed consent; the study was approved by the institutional review board of all participating centers, monitored by an independent data and safety monitoring board, and conducted in accordance with the principles of the Declaration of Helsinki. Merck & Co (Kenilworth, NJ) provided the study drug and study funding. Data collection and analysis were performed independently by the investigators with no involvement of the funding source.
Treatment, assessments, and end points
Patients had to have begun study treatment no later than 21 days from the post-ASCT discharge, recovered from ASCT toxicities at the time of first study treatment, and been no more than 60 days from stem cell reinfusion at study treatment start. All patients received up to 8 cycles of pembrolizumab administered at a flat dose of 200 mg IV every 3 weeks. Dose modification was not allowed. Doses could be delayed up to 12 weeks for toxicity. Drug was permanently held for grade 4 treatment-related adverse events (TRAEs), as well as for selected grade 3 immune-related AEs (irAEs). Safety was continuously monitored and graded using the Common Terminology Criteria for Adverse Events (CTCAE) version 4. Tumor assessments consisted of PET and computed tomography (CT) scans at baseline (BL; after ASCT and within 21 days prior to study treatment), at weeks 10 and 22 (before the fourth and eighth cycles, respectively) on treatment, and at 12 and 18 months post-ASCT. For patients with apparent radiographic progression of disease at week 10, continued treatment was allowed in cases where the patient was tolerating treatment without clinical deterioration. Radiographic assessments followed the International Harmonization Project 200713 criteria with all scans centrally reviewed in a dedicated imaging core facility. The primary end point was the PFS rate at 18 months after ASCT. Secondary end points included safety, 18-month overall survival (OS), PFS, and OS in the high-risk subset of patients not in complete remission (CR) prior to ASCT, and response rate to pembrolizumab in patients with measurable disease after ASCT, based on tumor imaging obtained at screening.
Statistical analysis
PFS and OS from the time of study entry were estimated as the proportion of evaluable patients alive and progression-free, or alive, respectively, at the 18-month time point. We hypothesized that pembrolizumab consolidation could improve the 18-month progression-free rate from a BL of 60%, estimated from historical results,14,15 to a rate of 80%. The null hypothesis of 60% was based using a lower bound of ∼55%, obtained from a large study of patients with induction failure or early relapse cHL2 (and therefore slightly higher expected risk than our unselected population) and using an upper bound of ∼65%, obtained from the transplant arm of a randomized study of chemosensitive patients undergoing ASCT that excluded patients with induction failure15 (and therefore slightly lower expected risk than our unselected population). With a planned sample size of 30 patients, the treatment would be considered promising if at least 22 of 30 patients remained alive and progression-free at 18 months. This design had a power of 87%, at a significance level of 0.09. PFS was also estimated using the Kaplan-Meier method. PFS was defined as the time from study entry to death from any cause, relapse, or progression, with patients censored at the last time seen alive and progression-free. OS was defined as the time from study entry to death from any cause, with patients censored at the last time seen alive. All calculations were performed using SAS 9.4 (SAS Institute Inc, Cary, NC).
Flow cytometry
Peripheral blood lymphocytes were analyzed using multiparameter flow cytometry. For comparison, we used a cohort of patients with cHL treated off-protocol at Dana-Farber Cancer Institute/Brigham and Women’s Hospital with ASCT using the same conditioning regimen, without PD-1 consolidation, in whom peripheral blood samples were collected, when possible, at similar time points post-ASCT. The protocol-specified time points were BL, then 3 weeks, 6 weeks, 9 weeks, 15 weeks, 21 weeks on treatment, then 12 and 18 months post-ASCT. For simplicity of analysis and reporting, we considered the following time points, all considered from the time of ASCT: study BL, then 2, 3, 6, 12, and 18 months. Samples were grouped with the closest specified time point. For each time point, the absolute number of circulating cells for a given subset was compared between study and control patients using Wilcoxon rank-sum testing; the significance level was set as 0.01 to account for multiplicity of testing. Whole blood (collected in 15% EDTA tubes) was incubated with a 10× volume of BDPharmlyse for 15 minutes at room temperature (RT) and subsequently centrifuged 1500 rpm for 10 minutes at RT. The cell pellet was resuspended in 500 μL of BDPharmlyse and aliquoted into 4 tubes for immunophenotypic analysis with the following panel of fluorophore-conjugated mAbs, including anti-CD3 V450 (clone UCHT1; BD Biosciences), anti-CD4 allophycocyanin (APC)-H7 (clone RPA-T4; BD Pharmingen), anti-CD8 Pacific Orange (clone RPA-T8; BioLegend), anti-CD25 phycoerythrin (PE)-Cy7 (clone M-A251; BD Pharmingen), anti-CD127 PE-Cy5 (clone eBioRDR5; eBioscience), anti-CD19 APC (clone HIB19; BD Pharmingen), and anti-CD56 PE (clone B159; BD Pharmingen) (among others in the full panel). Samples were incubated for 10 minutes with the respective mAb cocktail at RT in the dark and subsequently centrifuged 1500 rpm for 10 minutes at RT. Cell pellet was resuspended with 200 μL of BDPharmlyse. Samples were run in a 96-well plate on LSRFortessa (BD Biosciences) and data were analyzed with FACSDiva software (BD Biosciences).
Multiplex immunofluorescence
Archival formalin-fixed paraffin-embedded tissue was collected for all patients, when available. We accepted any pre-ASCT biopsy as “baseline,” whether obtained at the time of relapse or at the time of diagnosis; however, when available, we prioritized samples obtained at relapse for analysis. In addition, for patients who progressed after ASCT, a postprogression (PP) biopsy was requested whenever possible. Multiplex immunofluorescence staining was performed overnight for ∼12 hours on BOND RX fully automated stainers (Leica Biosystems). Tissue sections of 5-μm-thick formalin-fixed paraffin-embedded were baked for 3 hours at 60°C before loading into the BOND RX. Slides were deparaffinized (BOND DeWax Solution; Leica Biosystems) and rehydrated with series of graded ethanol to deionized water. Antigen retrieval was performed in BOND Epitope Retrieval Solution 1 (ER1; Leica Biosystems) at pH 6 for 10 minutes at 98°C. Deparaffinization, rehydration, and antigen retrieval were all preprogrammed and executed by the BOND RX. Next, slides were serially stained with primary antibodies. Incubation time per primary antibody was 40 minutes. Subsequently, anti-rabbit polymeric horseradish peroxidase (BOND Polymer Refine Detection kit; Leica Biosystems) was applied as a secondary label with an incubation time of 10 minutes. Signal for antibody complexes was labeled and visualized through corresponding tyramide-conjugated opal fluorophore reagents by incubating the slides for 5 minutes as previously described.6 The same process was repeated for subsequent antibodies/fluorescent dyes. Slides were air dried, mounted with Prolong Diamond Anti-fade mounting medium (Life Technologies) and stored in a lightproof box at 4°C prior to imaging. Image acquisition was performed using the Mantra multispectral imaging platform (Vectra 3; PerkinElmer, Hopkinton, MA). Areas with nontumor or residual normal tissue (ie, residual lymph node) were excluded from the analysis. Representative regions of interest were chosen by the pathologist (S.J.R.), and 3 to 5 fields of view were acquired at ×20 resolution as multispectral images. After image capture, the fields of view were spectrally unmixed and then analyzed using supervised machine-learning algorithms within Inform 2.4 (PerkinElmer). This image analysis software assigns phenotypes to all cells in the image, based on a combination of immunofluorescence characteristics associated with segmented nuclei (4′,6-diamidino-2-phenylindole [DAPI] signal). Each cell-phenotype–specific algorithm is based upon an iterative training/testing process, whereby a small number of cells are manually selected as being most representative of each phenotype of interest and the algorithm then predicts the phenotype for all remaining cells. Thresholds for positive staining and the accuracy of phenotypic algorithms were optimized and confirmed by the pathologist (S.J.R.) for each case.
Results
Patients
Thirty-one patients were enrolled on this study between April 2015 and March 2017; 1 patient withdrew consent prior to treatment and was replaced. The BL characteristics of the 30 eligible patients are shown in Table 1. The median age was 33 years (range, 20-69 years). The most common first-line therapy was doxorubicin, bleomycin, vinblastine, and dacarbazine (ABVD; 77% of patients), and the most common salvage therapy was ifosfamide, carboplatin, and etoposide (ICE; 50% of patients). Six patients (20%) had received PD-1 blockade as part of salvage therapy. All patients received carmustine, etoposide, cytarabine, and melphalan (BEAM) for ASCT conditioning. Using the recently described 5 risk factors for patients with cHL undergoing ASCT (primary refractory disease or relapse within 12 months of frontline therapy, residual fluorodeoxyglucose [FDG]-avid disease after salvage, >1 salvage regimen needed to achieve remission, extranodal disease at relapse, or B symptoms at relapse),3 90% of patients had at least 1 risk factor, and 40% of patients had 2 or more; 26 patients (87%) would have met eligibility criteria for the high-risk AETHERA study14 (primary refractory disease, relapse within 12 months, or extranodal disease at relapse).
Baseline patient characteristics
| Variable | N (%*) or median (range) |
|---|---|
| Total | 30 (100) |
| Age, y | 33 (20-69) |
| Sex | |
| Male | 16 (53) |
| Female | 14 (47) |
| Frontline therapy | |
| A(B)VD† | 24 (80) |
| BV-A(B)VD | 1 (3) |
| ABVE-PC | 1 (3) |
| BEACOPP‡ | 2 (7) |
| RCHOP/REPOCH | 2 (7) |
| Prior brentuximab exposure | 6 (20) |
| Prior nivolumab or pembrolizumab exposure | 6 (20) |
| Prior radiotherapy | 7 (23) |
| Conditioning regimen | |
| BEAM | 30 (100) |
| Risk factors | |
| Primary refractory disease | 17 (57) |
| Relapse within 12 mo | 5 (17) |
| Extranodal disease at relapse | 8 (27) |
| At least 1 of above 3 factors | 26 (87) |
| Residual disease after salvage | 3 (10) |
| B symptoms at relapse | 2 (7) |
| >1 salvage therapy | 5 (17) |
| At least 1 of above 6 factors | 27 (90) |
| At least 2 of above 6 factors | 12 (40) |
| Disease status at study entry (post-ASCT) | |
| Partial remission | 2 (7) |
| Complete remission | 28 (93) |
| Variable | N (%*) or median (range) |
|---|---|
| Total | 30 (100) |
| Age, y | 33 (20-69) |
| Sex | |
| Male | 16 (53) |
| Female | 14 (47) |
| Frontline therapy | |
| A(B)VD† | 24 (80) |
| BV-A(B)VD | 1 (3) |
| ABVE-PC | 1 (3) |
| BEACOPP‡ | 2 (7) |
| RCHOP/REPOCH | 2 (7) |
| Prior brentuximab exposure | 6 (20) |
| Prior nivolumab or pembrolizumab exposure | 6 (20) |
| Prior radiotherapy | 7 (23) |
| Conditioning regimen | |
| BEAM | 30 (100) |
| Risk factors | |
| Primary refractory disease | 17 (57) |
| Relapse within 12 mo | 5 (17) |
| Extranodal disease at relapse | 8 (27) |
| At least 1 of above 3 factors | 26 (87) |
| Residual disease after salvage | 3 (10) |
| B symptoms at relapse | 2 (7) |
| >1 salvage therapy | 5 (17) |
| At least 1 of above 6 factors | 27 (90) |
| At least 2 of above 6 factors | 12 (40) |
| Disease status at study entry (post-ASCT) | |
| Partial remission | 2 (7) |
| Complete remission | 28 (93) |
A(B)VD, doxorubicin, bleomycin, vinblastine, and dacarbazine; ABVE-PC, doxorubicin, bleomycin, vincristine, etoposide, prednisone, cyclophosphamide; BEACOPP, bleomycin, etoposide, doxorubicin, cyclophosphamide, vincristine, procarbazine, and prednisone; BEAM, carmustine, etoposide, cytarabine, and melphalan; BV, brentuximab vedotin; RCHOP, rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone; REPOCH, rituximab plus etoposide, prednisone, vincristine, cyclophosphamide, and doxorubicin.
Percentages may not add to 100 because of rounding.
With rituximab in 1 patient.
Given after ABVD in 1 additional patient.
Patients began pembrolizumab at a median of 34 days (range, 27-43 days) from the time of stem cell reinfusion. Twenty-three patients (77%) completed all planned 8 cycles of therapy. Of the 7 who did not, 2 stopped for patient choice, 4 for toxicity, and 1 for progressive disease prior to the eighth cycle. Two patients had progressive disease at the first restaging before cycle 4, with subsequent confirmed progressive disease despite continued treatment. Two patients were lost to follow-up, both of whom were alive and in CR at 12 months post-ASCT.
Toxicity
Toxicity, including TRAEs and irAEs, is summarized in Table 2. Overall, 24 patients (80%) experienced at least 1 grade 2 or higher AE, and 9 (30%) at least 1 grade 3 or higher AE. Considering only AEs at least possibly related to study treatment, 15 patients (50%) had at least 1 grade 2 or higher TRAE, and 8 (27%) at least 1 grade 3 or higher TRAE. In total, there were 14 grade 3 TRAEs and 6 grade 4 TRAEs. There were no grade 5 AEs on this study. Considering only irAEs, there were 39 events in total, affecting 13 patients (43%). The most common grade 2 or higher irAE was pneumonitis, cough, or dyspnea (4 grade 2 and 1 grade 3 events). There were no grade 4 or higher irAEs. Five AEs led to discontinuation (1 by patient choice, 4 per protocol): grade 2 pneumonitis (n = 1), grade 2 diplopia (n = 1), grade 3 pneumonitis (n = 1), grade 3 transaminitis (n = 2).
Summary of toxicity
| AE | Grade 1 | Grade 2 | Grade 3 | Grade 4 |
|---|---|---|---|---|
| Total no. of AEs | 308 (93) | 96 (77) | 21 (30) | 7 (10) |
| Grade 2-4 TRAE | ||||
| Leukopenia | 4 (7) | 5 (13) | 1 (3) | |
| Neutropenia | 3 (7) | 4 (10) | ||
| Transaminitis | 3 (10) | 3 (7) | ||
| Diarrhea/colitis | 1 (3) | 3 (7) | ||
| Pneumonitis/dyspnea | 3 (7) | 1 (3) | ||
| Hypothyroidism | 4 (7) | |||
| Rash | 3 (7) | |||
| Lymphopenia | 1 (3) | 1 (3) | ||
| Thrombocytopenia | 2 (7) | |||
| Febrile neutropenia | 1 (3) | |||
| Pulmonary hemorrhage* | 1 (3) | |||
| Hyperthyroidism | 1 (3) | |||
| Arthritis | 1 (3) | |||
| Fatigue | 1 (3) | |||
| Neck pain | 1 (3) | |||
| Creatinine increase | 1 (3) | |||
| Blurred vision | 1 (3) | |||
| Total no. of TRAEs | 82 (67) | 30 (47) | 14 (27) | 6 (10) |
| Grade 2-4 irAEs | ||||
| Transaminitis | 2 (7) | 3 (7) | ||
| Pneumonitis/dyspnea/cough | 4 (10) | 1 (3) | ||
| Diarrhea/colitis | 1 (3) | 2 (7) | ||
| Rash | 3 (7) | |||
| Hypothyroidism | 3 (3) | |||
| Pulmonary hemorrhage* | 1 (3) | |||
| Hyperthyroidism | 1 (3) | |||
| Arthritis | 1 (3) | |||
| Creatinine increase | 1 (3) | |||
| Total no. of irAEs | 16 (33) | 16 (33) | 7 (20) | 0 (0) |
| AE | Grade 1 | Grade 2 | Grade 3 | Grade 4 |
|---|---|---|---|---|
| Total no. of AEs | 308 (93) | 96 (77) | 21 (30) | 7 (10) |
| Grade 2-4 TRAE | ||||
| Leukopenia | 4 (7) | 5 (13) | 1 (3) | |
| Neutropenia | 3 (7) | 4 (10) | ||
| Transaminitis | 3 (10) | 3 (7) | ||
| Diarrhea/colitis | 1 (3) | 3 (7) | ||
| Pneumonitis/dyspnea | 3 (7) | 1 (3) | ||
| Hypothyroidism | 4 (7) | |||
| Rash | 3 (7) | |||
| Lymphopenia | 1 (3) | 1 (3) | ||
| Thrombocytopenia | 2 (7) | |||
| Febrile neutropenia | 1 (3) | |||
| Pulmonary hemorrhage* | 1 (3) | |||
| Hyperthyroidism | 1 (3) | |||
| Arthritis | 1 (3) | |||
| Fatigue | 1 (3) | |||
| Neck pain | 1 (3) | |||
| Creatinine increase | 1 (3) | |||
| Blurred vision | 1 (3) | |||
| Total no. of TRAEs | 82 (67) | 30 (47) | 14 (27) | 6 (10) |
| Grade 2-4 irAEs | ||||
| Transaminitis | 2 (7) | 3 (7) | ||
| Pneumonitis/dyspnea/cough | 4 (10) | 1 (3) | ||
| Diarrhea/colitis | 1 (3) | 2 (7) | ||
| Rash | 3 (7) | |||
| Hypothyroidism | 3 (3) | |||
| Pulmonary hemorrhage* | 1 (3) | |||
| Hyperthyroidism | 1 (3) | |||
| Arthritis | 1 (3) | |||
| Creatinine increase | 1 (3) | |||
| Total no. of irAEs | 16 (33) | 16 (33) | 7 (20) | 0 (0) |
“Treatment-related” refers to AEs judged to be at least possibly related to study treatment. The number of patients, expressed as the percentage of total patients, is given in parenthesis for grade 2-4 TRAEs. Events are ordered by frequency.
In a patient with prior cavitary tumor lesion.
Efficacy
Two patients had residual disease after ASCT as detected on their BL PET-CT prior to study initiation. Both were in CR after 3 cycles of pembrolizumab. One remained in CR after 18 months, and the other was in CR at 12 months and then lost to follow-up. At 12 months, 26 patients (87%) were in CR. By the 18-month time point (primary end point), 5 patients (17%) had relapsed, at a median of 6 months (range, 3-18 months) after ASCT. All others (83%) remained in CR, including the 2 patients in CR at 12 months who were lost to follow-up. The study therefore met its primary end point. Even if the 2 patients lost to follow-up at 12 months were considered PFS events, the 18-month progression-free rate (77%) would still have met the primary end point. The Kaplan-Meier estimate of 19-month PFS (using a 19-month time point to account for the variability of the exact time of the “18-month” restaging) was 81% overall (95% confidence interval [95CI], 60% to 92%) (Figure 1A). Among high-risk patients with at least 1 of the 5 aforementioned risk factors, the 19-month PFS was 85% (95CI, 64% to 94%) (Figure 1B). Among patients with at least 2 high-risk factors, it was 83% (95CI, 48% to 96%). Among patients who would have been eligible for the AETHERA study (primary refractory disease or relapse within 12 months, or extranodal disease at relapse), the 19-month PFS was 85% (95CI, 64% to 94%). The 19-month OS for the entire cohort was 100%.
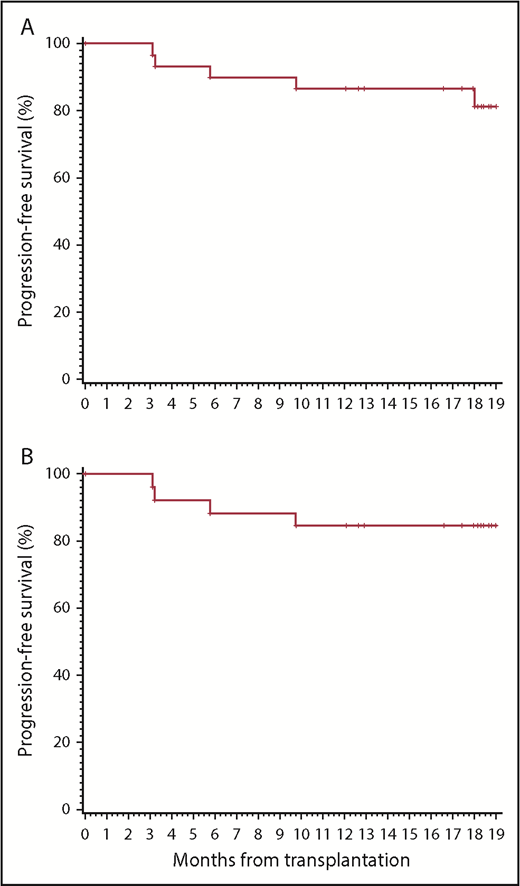
PFS. (A) All patients on study. (B) Patients with at least 1 clinical risk factor (primary refractory or relapse within 1 year, residual FDG-avid disease at ASCT, >1 salvage regimen, extranodal disease or B symptoms at relapse).
PFS. (A) All patients on study. (B) Patients with at least 1 clinical risk factor (primary refractory or relapse within 1 year, residual FDG-avid disease at ASCT, >1 salvage regimen, extranodal disease or B symptoms at relapse).
Correlative studies
Immune reconstitution post-ASCT was examined with flow cytometry of peripheral blood mononuclear cells. For this, we compared the absolute and relative amounts of various lymphocyte subsets between the study patients and a cohort of control patients (see “Methods”). There were 30 control patients with available samples. Their median age (32 years; range, 21-63 years) and sex (60% male) were similar to study patients. With adjustment for multiple comparisons, we found no significant difference in total CD4+ or CD8+ T cells, regulatory T cells, natural killer (NK) cells, or B cells in the pembrolizumab-treated patients (Table 3). However, there was a trend toward a higher number of NK cells at 3 and 6 months after ASCT in the pembrolizumab-treated patients.
Immune reconstitution
| Lymphocyte subset | Time point post-ASCT* | |||||
|---|---|---|---|---|---|---|
| BL, 0-45 d post-ASCT | 2 mo | 3 mo | 6 mo | 12 mo | 18 mo | |
| Effector T cells (CD3+CD8+) | ||||||
| Study patients† | 362.4 | 378.5 | 421.7 | 389.4 | 380.9 | 248.3 |
| Controls† | 260.4 | 372.5 | 196.5 | 189.2 | 222.8 | 250.8 |
| P | .16 | .41 | .056 | .11 | .37 | .37 |
| Helper T cells (CD3+CD4+) | ||||||
| Study patients† | 200.1 | 217.5 | 196.6 | 193.9 | 227.9 | 288.5 |
| Controls† | 290.4 | 267.6 | 181.4 | 222 | 288.5 | 319.3 |
| P | .16 | .49 | .31 | .62 | .22 | .63 |
| Regulatory T cells (CD4+CD25+CD127−) | ||||||
| Study patients† | 24.6 | 26.6 | 19.5 | 18.6 | 19.1 | 27.2 |
| Controls† | 35 | 11.1 | 14.3 | 11.7 | 17.6 | 21.5 |
| P | .52 | .078 | .15 | .036 | .87 | .16 |
| NK cells (CD3−CD56+) | ||||||
| Study patients† | 260.1 | 192 | 148.6 | 152.7 | 97.8 | 130.5 |
| Controls† | 199.4 | 169.2 | 89.7 | 71.2 | 117.4 | 110.5 |
| P | .081 | .82 | .026 | .026 | .84 | .82 |
| B cells (CD19+) | ||||||
| Study patients† | 7.8 | 131.4 | 155.1 | 190.9 | 275.1 | 269.9 |
| Controls† | 3 | 143.9 | 32.3 | 207.5 | 297.2 | 201.3 |
| P | .12 | .49 | .022 | .73 | .7 | .94 |
| Lymphocyte subset | Time point post-ASCT* | |||||
|---|---|---|---|---|---|---|
| BL, 0-45 d post-ASCT | 2 mo | 3 mo | 6 mo | 12 mo | 18 mo | |
| Effector T cells (CD3+CD8+) | ||||||
| Study patients† | 362.4 | 378.5 | 421.7 | 389.4 | 380.9 | 248.3 |
| Controls† | 260.4 | 372.5 | 196.5 | 189.2 | 222.8 | 250.8 |
| P | .16 | .41 | .056 | .11 | .37 | .37 |
| Helper T cells (CD3+CD4+) | ||||||
| Study patients† | 200.1 | 217.5 | 196.6 | 193.9 | 227.9 | 288.5 |
| Controls† | 290.4 | 267.6 | 181.4 | 222 | 288.5 | 319.3 |
| P | .16 | .49 | .31 | .62 | .22 | .63 |
| Regulatory T cells (CD4+CD25+CD127−) | ||||||
| Study patients† | 24.6 | 26.6 | 19.5 | 18.6 | 19.1 | 27.2 |
| Controls† | 35 | 11.1 | 14.3 | 11.7 | 17.6 | 21.5 |
| P | .52 | .078 | .15 | .036 | .87 | .16 |
| NK cells (CD3−CD56+) | ||||||
| Study patients† | 260.1 | 192 | 148.6 | 152.7 | 97.8 | 130.5 |
| Controls† | 199.4 | 169.2 | 89.7 | 71.2 | 117.4 | 110.5 |
| P | .081 | .82 | .026 | .026 | .84 | .82 |
| B cells (CD19+) | ||||||
| Study patients† | 7.8 | 131.4 | 155.1 | 190.9 | 275.1 | 269.9 |
| Controls† | 3 | 143.9 | 32.3 | 207.5 | 297.2 | 201.3 |
| P | .12 | .49 | .022 | .73 | .7 | .94 |
Median absolute numbers of selected lymphocyte subsets are shown post-ASCT and compared between study patients (N = 30; treated with pembrolizumab) and control patients (N = 30; no pembrolizumab) (see “Methods” for details). The significance level was set as 0.01 to account for multiplicity of testing.
Samples drawn outside of the given time points are grouped with the closest specified time point (see “Methods”). Not all patients had available samples at all time points.
Median of absolute numbers, in cells ×106/L.
Multiplex immunofluorescence analyses of BL (which could be archival, see “Methods”) and PP biopsies were conducted when available. Among the 5 patients with progression, 2 had tissue available for study. In 1 of them, no HRS cells could be definitively identified on morphological evaluation or by multiplex imaging. In the patient with available BL (which was obtained for this patient at the time of initial diagnosis) and PP biopsies, there was increased PD-L1 expression on macrophages between the 2 biopsies (18% of CD68+ cells were PD-L1+ at BL, vs 33% PP) and HRS cells (4% of HRS PD-L1+ at BL vs 34% PP) (Figure 2), as well as increased PD-L2 expression on HRS cells (50% HRS cells PD-L2+ at BL vs 90% PP). There was also slightly increased PD-1 expression (4% of CD3+ were cells PD-1+ at BL vs 11% PP) on infiltrating T cells.
![Figure 2. Multiplex immunofluorescence. Comparison of BL and PP biopsy for patient with available tissue. (A) Increased PD-L1 on HRS (PAX5+, red arrows) cells and macrophages (CD68+). White arrow indicates PAX5+ B cell. Original magnification ×200; inset, original magnification ×200. (B) Increase in PD-1 on infiltrating T cells (CD3+). Original magnification ×200; inset, original magnification ×200. Stains: DAPI (Invitrogen); PDL1: 9A11 (Dana-Farber Cancer Institute [DFCI]/Cell Signaling Technology [CST]), Opal 520; PD1: EH33 (DFCI/CST), Opal 690; CD3: PolyC (Dako), Opal 570; CD68: PGM1 (Dako), Opal 540; PAX5: 24/PAX-5 (BD Transduction), Opal 620.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/134/1/10.1182_blood.2019000215/3/m_bloodbld2019000215f2.png?Expires=1770589503&Signature=LcFOcf~7H~3lhNu--KJGQ1oTvWLcpVJPbKpT8nGUz6eeds4WkOIaxnqTHsi6QQhOz-xuC30SawjrPun~yUOrmMprftYIPHUStRX0h94TKpZav811p-qHVenSlSy2J~KNI6g-pNH7v9Nz~kUTGgpssmsr6YK1WUe502g0bmUHeOuv9VvTTXXiRXVjsL-VPNVGeyaVNzuqSKSKCtJjjiKFS-T4Z6KNe-iW34S4Tw6iDTRb2bTB51-ZKYS5Tx28VrXOjYiMoPRrLQtiz3PfzNyXvnnZUWFK9xeEt47nchuUGb5WFcLrU9PcfLpZ77Sx--9dX3UrJJRQbjGEiP~bJhZwhA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Multiplex immunofluorescence. Comparison of BL and PP biopsy for patient with available tissue. (A) Increased PD-L1 on HRS (PAX5+, red arrows) cells and macrophages (CD68+). White arrow indicates PAX5+ B cell. Original magnification ×200; inset, original magnification ×200. (B) Increase in PD-1 on infiltrating T cells (CD3+). Original magnification ×200; inset, original magnification ×200. Stains: DAPI (Invitrogen); PDL1: 9A11 (Dana-Farber Cancer Institute [DFCI]/Cell Signaling Technology [CST]), Opal 520; PD1: EH33 (DFCI/CST), Opal 690; CD3: PolyC (Dako), Opal 570; CD68: PGM1 (Dako), Opal 540; PAX5: 24/PAX-5 (BD Transduction), Opal 620.
Multiplex immunofluorescence. Comparison of BL and PP biopsy for patient with available tissue. (A) Increased PD-L1 on HRS (PAX5+, red arrows) cells and macrophages (CD68+). White arrow indicates PAX5+ B cell. Original magnification ×200; inset, original magnification ×200. (B) Increase in PD-1 on infiltrating T cells (CD3+). Original magnification ×200; inset, original magnification ×200. Stains: DAPI (Invitrogen); PDL1: 9A11 (Dana-Farber Cancer Institute [DFCI]/Cell Signaling Technology [CST]), Opal 520; PD1: EH33 (DFCI/CST), Opal 690; CD3: PolyC (Dako), Opal 570; CD68: PGM1 (Dako), Opal 540; PAX5: 24/PAX-5 (BD Transduction), Opal 620.
Discussion
This is, to our knowledge, the first study of immune-checkpoint blockade used for post-ASCT consolidation in patients with RR cHL. The AETHERA study demonstrated the benefit of BV used in a similar setting, with a significant PFS benefit.3 The continued improvement in PFS at 5 years in patients who received BV consolidation raises the possibility that this strategy can improve the cure rate of ASCT. This suggests more generally that treatments that by themselves may not have strong curative potential in advanced RR disease can, in the context of ASCT, be delivered with curative intent. Given that PD-1 blockade in RR cHL is associated with high response rates (∼70%) and durable responses (∼18 months),16,17 its use post-ASCT may similarly be associated with an improvement in long-term PFS that could translate to an increase in cure rate. The present study demonstrates the feasibility of this strategy. The toxicity of pembrolizumab in this setting appeared similar to its use in other settings, with a frequency of severe TRAEs that mirrored the frequency in the phase 2 KEYNOTE-087 study,10 and no unexpected severe AEs. Some toxicities, especially cytopenias and pneumonitis, are expected after ASCT using BEAM conditioning. Despite this, the rate of severe pneumonitis was low with only 1 grade 3 event, and the rate of severe cytopenias was also low. In addition, pembrolizumab did not significantly perturb the immune reconstitution of patients after ASCT. We did find a trend toward a possible earlier NK recovery in the pembrolizumab-treated patients. This finding, which should be validated, is noteworthy given the growing recognition of the potential importance of NK cells in cHL. NK-cell abundance and activation status may be altered in cHL and modulated by PD-1 blockade18 ; NK cells may be recruited to the vicinity of HRS with therapeutic benefit19 ; and responses to PD-1 blockade may occur in the absence of functional class I and class II major histocompatibility complex expression on tumor cells, suggesting that NK cells may participate in the antitumor activity of PD-1 blockade.20
The paucity of available PP biopsies did not allow us to formulate clear hypotheses for the mechanism of treatment failure. In the 1 sample that could be adequately evaluated, we observed robust PD-L1 or PD1 expression within the tumor microenvironment indicating that loss of these therapeutic targets was not associated with progressive disease. Rather, we observed an increase in PD-1 expression on tumor-infiltrating T cells and an increase in PD-L1 expression on HRS cells and PD-L1 expression on macrophages in the biopsy at progression compared with the BL biopsy sample. The relationship between PD-L1 expression on tumor cells and macrophages has previously been reported and suggests close cross-talk between the HRS cell and its immune microenvironment.6 The increase in PD-L1/PD-1 expression, which has been observed in other settings in cHL,21,22 suggests that the tumor may increase signaling through the PD-1 pathway as a broad mechanism of therapy resistance. It is interesting to observe this phenomenon even under PD-1 blockade. If confirmed, this finding raises the hypothesis that the increased PD-L1 expression seen in relapsing tumors is not due primarily to the selection of tumor subclones with higher expression; instead, it could be the result of tumor microenvironmental changes induced broadly by tumor-toxic treatments that result in increased PD-L1 expression in the HRS cells.
With an estimated 19-month PFS of 81%, the study met its primary end point. It is challenging to establish the relevant historical comparison PFS rate, given the difference in eligibility criteria and risk mix of this study compared with prior publications. The null hypothesis of 60% was based on 2 large historical series (see “Methods”). In addition, it may be useful to compare our results to those of the AETHERA study.3 This trial, which was not published at the time our study started, is one of the largest recent experiences of ASCT for RR cHL, specifically examining the impact of maintenance therapy (as did our study) and, most importantly, providing a modern reassessment of clinical high-risk factors that can be used to calibrate our observed PFS against expectations. Few patients in our trial had residual FDG-avid disease after salvage therapy, which by itself should translate to better post-ASCT PFS. However, almost all had at least 1 other clinical high-risk factor, including 87% who would have been eligible for the AETHERA study14 ; the 19-month PFS of 85% in this subgroup compares favorably to that of patients treated with placebo (∼45%) and that of patients treated with BV (∼70%) on AETHERA. We note the additional caveat that our study required patients to be in partial remission or CR after salvage and to have received no more than 2 lines of salvage, which were not eligibility criteria for AETHERA. The present results are also promising compared with a retrospective 2-institution study of patients who received transplants for early relapse or primary refractory cHL in which the 18-month event-free survival was <60%.2 Acknowledging the limitation of a small sample size, the PFS was high regardless of the number of risk factors. Larger studies will be necessary to tease out the relevant factors associated with prognosis in patients receiving consolidation pembrolizumab after ASCT. It is possible that the putative benefit of PD-1 blockade and the demonstrated benefit of BV could be combined by using both drugs together as consolidation; an ongoing study (NCT03057795) is testing this hypothesis. Ultimately, historical or cross-study comparisons are challenging, especially given the varying eligibility criteria and risk profiles, and the definitive demonstration of a significant benefit to PD-1 blockade consolidation must await the conduct of randomized studies. The present results strongly support the conduct of such studies, and raise the possibility that PD-1 blockade may be safely administered after ASCT and increase the cure rate for patients in whom frontline therapy is ineffective.
Individual participant data will not be shared. For original correlative data, please contact parmand@partners.org.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors are indebted to the nursing and research staff who were involved in this study, and to all of the participants and their families. The authors thank Mikel Lipschitz and the Center for Immuno-Oncology of the Dana-Farber Cancer Institute for help with immunostaining.
P.A. gratefully acknowledges the support of the Leukemia & Lymphoma Society (Scholar in Clinical Research), as well as the Harold and Virginia Lash Foundation, and the Pasquarello Tissue Bank in Hematologic Malignancies. A.F.H. was supported by the Lymphoma Research Foundation Larry and Denise Mason Clinical Investigator Career Development Award and the National Cancer Institute of the National Institutes of Health under award numbers 2K12CA001727 and P50CA107399. The study was funded by Merck and Co (Kenilworth, NJ).
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Authorship
Contribution: P.A. designed the research, collected and analyzed the data, and wrote the paper; Y.-B.C., R.M.J., J.B., E.J., K.C.C., P.B.D., Y.N., A.S.L., D.C.F., S.Y.N., O.O.O., A.S.F., A.I.K., J.L.C., C.A.J., E.D.J., and A.F.H. collected data and edited the paper; R.A.R. analyzed data and edited the paper; R.W.M., J.L.W., and S.S.P. collected and analyzed data and edited the paper; J.R. and S.J.R. designed the study, analyzed data, and edited the paper; and M.A.S. designed the study and edited the paper.
Conflict-of-interest disclosure: P.A. provided consulting services to Merck & Co, Bristol-Myers Squibb, Pfizer, Affimed, Adaptive, and Infinity, and received research funding (institutional) from Merck & Co, Bristol-Myers Squibb, Affimed, Adaptive, Roche, Tensha, Otsuka, and Sigma Tau. Y.-B.C. provided consulting services to Magenta, Takeda, Kiadis, Incyte, and Regimmune. D.C.F. received honoraria from Merck & Co. A.F.H. provided consulting services to Bristol-Myers Squibb, Genentech, Merck & Co, Kite Pharma/Gilead, Adaptive Biotechnologies, and Seattle Genetics, and received research funding from Bristol-Myers Squibb, Genentech, Immune Design, AstraZeneca, Merck & Co, Pharmacyclics, Seattle Genetics, Kite Pharma, and Gilead Sciences. C.A.J. provided consulting services to Kite Pharma, Novartis, Pfizer, Humanigen, Precision Bioscience, Bayer, and Celgene, and received research funding from Pfizer and Kite Pharma. E.D.J. provided consulting services to Merck & Co, AstraZeneca, and Seattle Genetics, and received research funding from Pharmacyclics and Celgene. A.S.L. provided consulting services to Bristol-Myers Squibb and received honoraria from, and served on speakers’ bureaus for, Seattle Genetics, Humanigen, and Research to Practice. J.R. provided consulting services to Avrobio, Celgene, Draper Labs, LifeVault Bio, Regenerex, and TScan Therapeutics, and received research funding from Prometheus Laboratories, Neovii Biotech, Nektar, Merck & Co, and Kite Pharma. Y.N. received research funding from Otsuka, AstraZeneca, Affimed, Celgene, Sanofi Aventis, and Novartis. S.J.R. received research funding from Bristol-Myers Squibb, Merck & Co, Kite Pharma/Gilead, and Affimed Pharmaceuticals. M.A.S. provided consulting services to Bristol-Myers Squibb and received research funding from Bayer, Bristol-Myers Squibb, and Merck & Co. The remaining authors declare no competing financial interests.
Correspondence: Philippe Armand, Dana-Farber Cancer Institute, 450 Brookline Ave, Boston MA 02215; e-mail: parmand@partners.org.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal