

In this edition of Blood, provide some new clues into how signals associated with infection and inflammation directly instruct hematopoietic stem cells (HSCs) to upregulate the myeloid differentiation program, which is required for a robust innate immune response.1
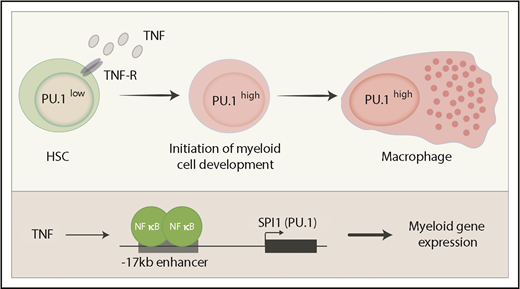
Induction of PU.1 by TNF. HSCs express relatively low amounts of PU.1 protein. Inflammatory pathways induce the expression of TNF, which directly activates SPI1 via the canonical NF-κB pathway. High PU.1 concentration then activates a large group of myeloid genes and promotes differentiation to mature myeloid lineages such as macrophages. TNF-R, TNF receptor.
Induction of PU.1 by TNF. HSCs express relatively low amounts of PU.1 protein. Inflammatory pathways induce the expression of TNF, which directly activates SPI1 via the canonical NF-κB pathway. High PU.1 concentration then activates a large group of myeloid genes and promotes differentiation to mature myeloid lineages such as macrophages. TNF-R, TNF receptor.
Hematopoiesis is a highly dynamic process that adjusts to the needs of the host in health (steady-state) as well as in response to emergencies such as infection or anemia.2 How the production of the major blood cell lineages from the HSCs is controlled to fulfill the various requirements of the host has been the subject of a considerable amount of research in the field over many decades. Two distinct paradigms have been proposed whose positions were captured by a mock debate in this journal 20 years ago between 2 of the main protagonists.3,4 The intrinsic model suggests that changes in gene regulation in HSCs, through the combined activity of major transcriptional regulators, result in conditions that favor one lineage fate over another.3,5 These intrinsic changes in transcription factor expression are often proposed to be stochastic in nature. In contrast, the extrinsic model suggests that extracellular signals, most notably cytokines, direct the cell fate of HSCs into particular lineages.4,6 Despite the simplicity of these 2 concepts, they have proven extremely difficult to test, partly because of the rarity of true HSCs, limitations when analyzing cells on a bulk population level, and redundancy in cytokine signals.
To investigate the differentiation of HSCs to lineage-restricted progeny, several studies, including that by Etzrodt et al, have tracked the expression of the major myeloid transcription factor PU.1 (encoded by the SPI1 gene). SPI1 is expressed in HSCs at a lower level before being upregulated in all myeloid cells, where it plays a major role in their differentiation and function.7,8 In contrast, SPI1 is downregulated or silenced completely in all lymphoid cells. Hence changes in PU.1 concentration serve as a useful and functionally important readout for the differentiation status of hematopoietic progenitors. Etzrodt et al used single-cell quantitative time lapse imaging of mouse HSCs carrying a fluorescently tagged PU.1 to systematically examine a panel of cytokines for their direct impact on PU.1 expression and thus hematopoietic differentiation. The authors tracked PU.1 concentration in >100 individual HSCs in each experimental condition and found 2 cytokines, interleukin-1β (IL-1β) and tumor necrosis factor (TNF), that induced PU.1 over a 12-hour time course. The induction of PU.1 occurred at the level of new messenger RNA transcription and TNF, at least, was demonstrated to signal through the canonical NF-κB pathway (see figure).
To examine whether these inflammatory cytokines induce PU.1 expression in vivo, Etzrodt et al made use of toll-like receptor ligands such as lipopolysaccharide (LPS) that robustly activate the expression of a suite of such cytokines. As expected, PU.1 concentration increased in HSCs from LPS-treated mice, a finding that was mimicked by the injection of only TNF. Strikingly, TNF deficiency blocked any LPS-induced change in PU.1, suggesting that TNF was both necessary and sufficient for PU.1 upregulation in both isolated HSCs in culture and in the complex environment of the host.
On a single HSC level, the upregulation of PU.1 was heterogeneous in all systems examined. This was not the result of variation in the differentiation state of individual progenitors, because subfractionation of the HSCs showed similar variability in PU.1 induction. The existence of heterogeneity in all HSC fractions may suggest that cell intrinsic mechanisms are also at play in this model.
The conclusions of Etzrodt et al differ from those in a previous study that showed a similar activity for colony stimulating factor 1 (CSF1; also known as macrophage colony-stimulating factor [M-CSF]) in upregulating PU.1 and myeloid differentiation,9 although neither study has directly compared CSF1 to TNF (or IL-1β). CSF1 is also robustly induced by LPS and promotes the differentiation of more mature myeloid cells, although Etzrodt et al suggest that CSF1 has its impact indirectly of HSCs, most likely through TNF. Future studies that address this issue more directly are already in progress. PU.1 protein has also been proposed to accumulate to high concentrations in developing myeloid cells as a consequence of the lengthening of the cell cycle in the context of constant transcription.10 Such a process is not necessarily incompatible with the cytokine induced differentiation model presented by Etzrodt et al, particularly in the context of proliferating progenitors downstream of the HSCs.
In summary, the study by Etzrodt et al proposes that TNF is one of the long postulated extrinsic regulators of cell fate decisions in hematopoiesis. However, it is important to note that most studies in this field, including the study by Etzrodt et al, by necessity measure inflammation- or stress-induced hematopoiesis, such as the response of LPS, a mimic of bacterial infection. Although this pathway is certainly important for host responses to pathogens and other inflammatory stresses, the factors that regulate the interplay between intrinsic and extrinsic regulators of steady-state hematopoiesis in a healthy individual remain to be revealed.
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal