

Key Points
White patients with HVLPD more often had earlier-onset disease, lower EBV DNA, and normal numbers of NK cells in the blood than nonwhites.
White patients with HVLPD were less likely to develop systemic disease or require hematopoietic stem cell transplant than nonwhites.

Abstract
Patients with classic hydroa vacciniforme–like lymphoproliferative disorder (HVLPD) typically have high levels of Epstein-Barr virus (EBV) DNA in T cells and/or natural killer (NK) cells in blood and skin lesions induced by sun exposure that are infiltrated with EBV-infected lymphocytes. HVLPD is very rare in the United States and Europe but more common in Asia and South America. The disease can progress to a systemic form that may result in fatal lymphoma. We report our 11-year experience with 16 HVLPD patients from the United States and England and found that whites were less likely to develop systemic EBV disease (1/10) than nonwhites (5/6). All (10/10) of the white patients were generally in good health at last follow-up, while two-thirds (4/6) of the nonwhite patients required hematopoietic stem cell transplantation. Nonwhite patients had later age of onset of HVLPD than white patients (median age, 8 vs 5 years) and higher levels of EBV DNA (median, 1 515 000 vs 250 000 copies/ml) and more often had low numbers of NK cells (83% vs 50% of patients) and T-cell clones in the blood (83% vs 30% of patients). RNA-sequencing analysis of an HVLPD skin lesion in a white patient compared with his normal skin showed increased expression of interferon-γ and chemokines that attract T cells and NK cells. Thus, white patients with HVLPD were less likely to have systemic disease with EBV and had a much better prognosis than nonwhite patients. This trial was registered at www.clinicaltrials.gov as #NCT00369421 and #NCT00032513.
Introduction
Hydroa vacciniforme–like lymphoproliferative disorder (HVLPD) is a chronic illness that presents with papulovesicular skin lesions in sun-exposed areas, particularly the face or dorsum of the hands.1-5 The vesicles evolve into hemorrhagic crusts and heal with scarring. Skin biopsy specimens show reticular degeneration of the epidermis and a dermal infiltrate of Epstein-Barr virus (EBV)–infected T or natural killer (NK) cells. Patients often present in childhood with skin lesions after sun exposure and may have high levels of EBV DNA in T or NK cells in the blood. Patients with the classic or typical form of HVLPD usually have no systemic symptoms, normal hematologic findings, and high levels of EBV DNA in the blood.6 Some children have complete resolution of the disease by adolescence.7 T cells in the lesions are usually polyclonal or oligoclonal. Patients frequently have increased numbers of γ-δ T cells in the peripheral blood, which are often EBV positive. The disease can regress in adulthood or progress to a more severe systemic form of HVLPD with skin lesions in non–sun-exposed areas, deep ulcers, hepatic failure, and malignant lymphoma. HVLPD is more common in Asia and Central and South America than the United States or Europe.
Patients with the systemic (or severe) form of HVLPD have systemic symptoms, including persistent fever, weight loss, facial edema, lesions in nonexposed skin, lymphadenopathy, hepatosplenomegaly, hepatitis, and leukopenia reflecting systemic involvement with chronic active EBV disease (CAEBV).2,8 Skin biopsy specimens often show diffuse, deep ulcerative lesions with cellular atypia,9 and patients have high levels of EBV DNA in the blood.6 Patients may progress to EBV-positive T- or NK-cell lymphoma/leukemia or hemophagocytic syndrome. T cells in lesions are often monoclonal. Some patients with HVLPD also have hypersensitivity to mosquito bites (HMB) with necrosis, elevated levels of immunoglobulin E (IgE), and increased numbers of EBV-positive NK cells in the peripheral blood.
Here, we report our 11-year experience in evaluating patients with HVLPD at the Clinical Center of the National Institutes of Health (NIH). We found that white patients generally had a benign course, while 4 of 6 nonwhite patients required hematopoietic stem cell transplantation (HSCT) for their disease.
Methods
Patients
All patients or their parents (in the case of minors) signed consents and were enrolled on protocols approved by the institutional review board of the National Institute of Allergy and Infectious Diseases and the National Human Genome Research Institute (www.clinicaltrials.gov #NCT00032513 and #NCT00369421, respectively). Patients with HVLPD were defined as having papulovesicular skin lesions in sun-exposed areas that healed with scarring and markedly elevated levels of EBV DNA (>10 000 copies/mL) in the blood with high levels of EBV DNA in T or NK cells. Patients 1,10 11,11 and 1312 were reported previously. Patients with classic HVLPD had no persistent systemic symptoms, lymphadenopathy, hepatosplenomegaly, hepatitis, hemophagocytic syndrome, or NK-cell lymphocytosis, while those with systemic HVLPD had ≥1 of these persistent symptoms or signs of extracutaneous disease (based on prior criteria6 ).
Cell type of EBV DNA in blood, viral gene expression, and EBV typing
Peripheral blood mononuclear cells (PBMCs) were sorted using antibody to B, T, and NK cells. PBMCs were labeled with CD3-AF488, CD19-AF647, and CD56-phycoerythrin, and the cells were sorted by flow cytometry into T, B, and NK populations. The cells were lysed, real-time polymerase chain reaction (PCR) was performed using EBV DNA primers, and the number of EBV DNA copies per million cells was determined. Total RNA was isolated from PBMCs, complementary DNA was produced, and PCR was performed for EBV EBNA1, EBNA2, LMP1, and BZLF1 followed by Southern blotting.13,14 EBV type 1 or type 2 was determined by PCR.15 See supplemental Methods (available on the Blood Web site) for more information.
Results
Clinical findings and pathology of patients with HVLPD
Sixteen patients with HVLPD were evaluated at the NIH Clinical Center; 10 were white, 4 were Hispanic, and 2 were Asian (Table 1). All patients had skin lesions in sun-exposed areas, and some had lesions involving the conjunctiva (Figure 1). Pathology showed intraepidermal vesicles with reticular degeneration and necrotic epidermal keratinocytes with a mixture of inflammatory cells. Perivascular, periadnexal, and interstitial mononuclear inflammatory cells involved the full thickness of the dermis (Figure 2A). Thirteen patients were from the United States and 3 from Great Britain (patients 8-10). The median age at onset of disease was 5 years; white patients had a median age of onset of 5 years and nonwhite patients of 8 years. Patients with systemic HVLPD (persistent signs and symptoms with extracutaneous disease) had the same median age at diagnosis (6 years) as those with classic hydroa vacciniforme (HV). One of 10 white patients had systemic disease; patient 5 had EBV lymphoproliferative disease of the gastrointestinal tract (Figure 2C-D), which resolved spontaneously without treatment. In contrast, nonwhite patients were more likely to develop systemic disease (5/6) than white patients (1/10) (P = .008). Eighty-three percent of nonwhite patients had systemic disease with persistent fever and involvement of the lymph nodes, gastrointestinal tract, lungs, pleura, muscle, or liver; 1 patient had a T-cell lymphoma. All patients had skin biopsies confirming the diagnosis of HVLPD (Table 2), except patients 8 to 10, who underwent provocation testing with a UV-A solar simulator and had erythematous or vesicular responses typical for HVLPD.
Clinical features of HVLPD patients
| Patient | Race/ sex | Age at onset | Age at last follow-up | EBV-related symptoms | Classification of HV | Additional clinical features | Treatment |
|---|---|---|---|---|---|---|---|
| 1 | WM | 3 | 24 | None | Classic | None | None |
| 2 | WF | 5 | 15 | None | Classic | None | None |
| 3 | WM | 4 | 12 | None | Classic | Urticaria pigmentosa | Hydroxychloroquine |
| 4 | WM | 7 | 19 | None | Classic | Tetralogy of Fallot | None |
| 5 | WM | 6 | 28 | GI LPD | Systemic | Moebius syndrome | Hydroxychloroquine |
| 6 | WF | 3 | 16 | None | Classic | None | None |
| 7 | WF | 5 | 12 | None | Classic | Branchial cleft cyst on neck | Topical steroid |
| 8 | WF | 7 | 19 | None | Classic | Severe adenovirus conjunctivitis | Hydroxychloroquine, valacyclovir, prednisone pulses |
| 9 | WF | 7 | 16 | None | Classic | None | Prednisone pulses, valacyclovir, TL-01 narrow band UV-B and UV-A desensitization |
| 10 | WF | 3 | 11 | None | Classic | None | Valacyclovir, TL-01 (narrow-band UV-B) desensitization |
| 11 | HM | 3 | 16 | Hepatitis, lymphadenopathy, fever | Systemic | None | HSCT from 5/6 matched related donor |
| 12 | HM | 39 | 47 | None | Classic | None | None |
| 13 | AF | 10 | 27 | GI disease, pleural, muscle disease; T cell lymphoma of lung | Systemic | GATA2 deficiency | Prednisone, etoposide, chemotherapy, haploidentical HSCT from sister |
| 14 | AM | 8 | 23 | Hepatitis, fever, fatigue; NK/T-cell lymphoma after HSCT | Systemic | Renal insufficiency | Anakinra, prednisone, haploidentical HSCT from sister |
| 15 | HF | 6 | 15 | Fever, delirium, lymphocytic meningitis* | Systemic | None | Ganciclovir, intrathecal methotrexate |
| 16 | HM | 6 | 13 | Fever, abdominal pain | Systemic | None | Hydroxychloroquine, thalidomide, valacyclovir, HSCT |
| Patient | Race/ sex | Age at onset | Age at last follow-up | EBV-related symptoms | Classification of HV | Additional clinical features | Treatment |
|---|---|---|---|---|---|---|---|
| 1 | WM | 3 | 24 | None | Classic | None | None |
| 2 | WF | 5 | 15 | None | Classic | None | None |
| 3 | WM | 4 | 12 | None | Classic | Urticaria pigmentosa | Hydroxychloroquine |
| 4 | WM | 7 | 19 | None | Classic | Tetralogy of Fallot | None |
| 5 | WM | 6 | 28 | GI LPD | Systemic | Moebius syndrome | Hydroxychloroquine |
| 6 | WF | 3 | 16 | None | Classic | None | None |
| 7 | WF | 5 | 12 | None | Classic | Branchial cleft cyst on neck | Topical steroid |
| 8 | WF | 7 | 19 | None | Classic | Severe adenovirus conjunctivitis | Hydroxychloroquine, valacyclovir, prednisone pulses |
| 9 | WF | 7 | 16 | None | Classic | None | Prednisone pulses, valacyclovir, TL-01 narrow band UV-B and UV-A desensitization |
| 10 | WF | 3 | 11 | None | Classic | None | Valacyclovir, TL-01 (narrow-band UV-B) desensitization |
| 11 | HM | 3 | 16 | Hepatitis, lymphadenopathy, fever | Systemic | None | HSCT from 5/6 matched related donor |
| 12 | HM | 39 | 47 | None | Classic | None | None |
| 13 | AF | 10 | 27 | GI disease, pleural, muscle disease; T cell lymphoma of lung | Systemic | GATA2 deficiency | Prednisone, etoposide, chemotherapy, haploidentical HSCT from sister |
| 14 | AM | 8 | 23 | Hepatitis, fever, fatigue; NK/T-cell lymphoma after HSCT | Systemic | Renal insufficiency | Anakinra, prednisone, haploidentical HSCT from sister |
| 15 | HF | 6 | 15 | Fever, delirium, lymphocytic meningitis* | Systemic | None | Ganciclovir, intrathecal methotrexate |
| 16 | HM | 6 | 13 | Fever, abdominal pain | Systemic | None | Hydroxychloroquine, thalidomide, valacyclovir, HSCT |
A, Asian; F, female; GI, gastrointestinal; H, Hispanic; LPD, lymphoproliferative disease; M, male; W, white.
This patient developed systemic disease long after the onset of HV (9 years) manifested by lymphocytic meningitis; unlike the other patients with systemic disease, this patient has indeterminate T-cell clonality, which may explain the indolent course of her disease.

HV lesions. (A-G) Varioliform erosions on sun-exposed areas on the face of patient 4 (A), eroded papulopustules on the back of the neck of patient 1 (B), conjunctival involvement in patient 1 (C), erosions on the pinna of the ear of patient 5 (D), vesicles on the lip of patient 4 (E), bulla and healing erosions on the hand of patient 3 (F), and erosions on the neck of patient 4 (G). (H) Typical, round, punched out, varioliform scarring after healing of HV lesions on the cheek of patient 9.
HV lesions. (A-G) Varioliform erosions on sun-exposed areas on the face of patient 4 (A), eroded papulopustules on the back of the neck of patient 1 (B), conjunctival involvement in patient 1 (C), erosions on the pinna of the ear of patient 5 (D), vesicles on the lip of patient 4 (E), bulla and healing erosions on the hand of patient 3 (F), and erosions on the neck of patient 4 (G). (H) Typical, round, punched out, varioliform scarring after healing of HV lesions on the cheek of patient 9.
![Figure 2. Pathology of HV biopsy specimens. (A-B) Skin biopsy specimen from patient 5 showing multiloculated intraepidermal vesicles with reticular degeneration and necrotic epidermal keratinocytes and a mixed inflammatory reaction with neutrophils and mononuclear cells; no viral cytopathic changes were observed. The dermis showed perivascular, periadnexal, and interstitial mononuclear cells with a predominance of small lymphocytes throughout the thickness of the dermis (hematoxylin and eosin stain [(B) higher power]). (C-D) Terminal ileum and Peyer’s patches with reactive follicle (C; hematoxylin and eosin stain) and numerous EBV-positive cells (D; in situ hybridization for Epstein-Barr encoded RNA) in patient 5. (E-F) Skin biopsy specimen from patient 16 stained for CD3 (E) and Epstein-Barr encoded RNA (F; EBER). Original magnification ×100 (A,C), ×200 (D-F), ×400 (B).](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/133/26/10.1182_blood.2018893750/3/m_blood893750f2.png?Expires=1767709475&Signature=voCiMRLOixR4-2GkMgQuhJqav5i96WzoBflT-Z1ucbGxkR~LQMO-Akf32CD~L0wyh8pNIM896DqXuBirlu5WCar2euHFb8EFfhOF-jXdNM7OpL9DRx~tMe5D-qWvJ71eeiM--Ue9wF1AX7tHgz7puvpIelXadBR7QNs18NMkGHJ-GcdvLI41AXqnWiYxtqpBkOWDlramPIoRvnwd0kg82JKwGPKFsAV4FPxChzyToaVvaeJZcMOXXzyb5MVRcMwbNL83ROCCxT~RHdO6UsBtuzU7iu3-G32hdQnhOjfQs6lKPfo~1OALUI~Dnqa97qRRkRp7Hz5xmSGEORwQutr4-Q__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Pathology of HV biopsy specimens. (A-B) Skin biopsy specimen from patient 5 showing multiloculated intraepidermal vesicles with reticular degeneration and necrotic epidermal keratinocytes and a mixed inflammatory reaction with neutrophils and mononuclear cells; no viral cytopathic changes were observed. The dermis showed perivascular, periadnexal, and interstitial mononuclear cells with a predominance of small lymphocytes throughout the thickness of the dermis (hematoxylin and eosin stain [(B) higher power]). (C-D) Terminal ileum and Peyer’s patches with reactive follicle (C; hematoxylin and eosin stain) and numerous EBV-positive cells (D; in situ hybridization for Epstein-Barr encoded RNA) in patient 5. (E-F) Skin biopsy specimen from patient 16 stained for CD3 (E) and Epstein-Barr encoded RNA (F; EBER). Original magnification ×100 (A,C), ×200 (D-F), ×400 (B).
Pathology of HV biopsy specimens. (A-B) Skin biopsy specimen from patient 5 showing multiloculated intraepidermal vesicles with reticular degeneration and necrotic epidermal keratinocytes and a mixed inflammatory reaction with neutrophils and mononuclear cells; no viral cytopathic changes were observed. The dermis showed perivascular, periadnexal, and interstitial mononuclear cells with a predominance of small lymphocytes throughout the thickness of the dermis (hematoxylin and eosin stain [(B) higher power]). (C-D) Terminal ileum and Peyer’s patches with reactive follicle (C; hematoxylin and eosin stain) and numerous EBV-positive cells (D; in situ hybridization for Epstein-Barr encoded RNA) in patient 5. (E-F) Skin biopsy specimen from patient 16 stained for CD3 (E) and Epstein-Barr encoded RNA (F; EBER). Original magnification ×100 (A,C), ×200 (D-F), ×400 (B).
Virologic Features of HVLPD Patients
| Patient | EBV type | Sites of EBV+ cells | EBV cell type | First EBV load (copies/mL) | EBV gene expression | EBV copies/106 T/B/NK cells |
|---|---|---|---|---|---|---|
| 1 | 1 | Bone marrow EBV+, CSF+; skin EBV+ HVLPD | T cell | 403 000 | Negative for EBNA1, BZLF1 | 23 400 T/ 5356 B |
| 2 | ND | Skin HVLPD, EBV ND | T∼B cell | 94 850 | ND | 249 T/ 451 B |
| 3 | 1 | Skin EBV+ HVLPD, bone marrow EBV+ | T cell | 4 510 000 | Negative for EBNA1, BZLF1 | 350 638 T/ 12 148 B |
| 4 | 1 | skin HVLPD, EBV ND | T cell | 583 500 | Negative for EBNA1, BZLF1 | 486 661 T/ 6966 B |
| 5 | ND | Skin HVLPD, EBV ND; ileum EBV+ suggestive of T-cell LPD | T cell | 196 500 | ND | ND |
| 6 | 1 | Skin EBV+ HVLPD | T cell | 728 500 | Positive for EBNA1, BZLF1 (3+), Neg for LMP1, EBNA2 | 250 616 T/ 20 000 B |
| 7 | 1 | EBV+ HVLPD | T cell | 250 150 | Positive for EBNA1; Negative for EBNA2, LMP1, BZLF1 | 199 050 T/ 1761 B |
| 8 | ND | ND | T>NK | 83 180* | ND | 227 227 T/313 B/13 514 NK |
| 9 | 1 | ND | NK∼T | 100 000* | ND | 93 343 T/958 B/113 907 NK |
| 10 | 1 | ND | NK>T | 245 500* | ND | 43 005 T/857 B/245 979 NK |
| 11 | 1 | Skin T cell EBV+ HVLPD, EBV+ lymph node | T cell | 320 000† | Positive for EBNA1, LMP1, BZLF1 (1+), Neg for EBNA2 | ND |
| 12 | 2 | Skin HVLPD, EBV ND | T cell | 840 350 | Positive for EBNA1, BZLF1 (3+); Negative for LMP1, EBNA2 | 452 353 T/227 778 B |
| 13 | 1 | Skin, colon, pleura, muscle EBV+ HVLPD | B>T cell | 1 514 800 | Negative for EBNA1, BZLF1 | 2 022 000 T/18 410 000 B (EBV was predominantly in T cells in the tissues) |
| 14 | 1 | Skin (EBV+ HVLPD), liver, bone marrow, lymph node | T>NK cell | 3 630 780* | Positive for EBNA1; Negative for BZLF1, EBNA2, LMP1 | 5 843 000 T/84 000 B/310 000 NK |
| 15 | 1 | Skin (EBV+ HVLPD), bone marrow, buccal and lingual mucosa,CSF with lymphocytic pleocytosis | T∼NK cell | 1 659 586* | Positive for EBNA1; Negative for BZLF1, EBNA2, LMP1 | 1 633 000 T/20 000 B/1 513 000 NK |
| 16 | ND | Skin EBV+ HVLPD, lung and ileum EBV+ T cell lymphoma | T>NK cell | 3 388 442* | ND | 872 000 T/3672 B/660 000 NK |
| Patient | EBV type | Sites of EBV+ cells | EBV cell type | First EBV load (copies/mL) | EBV gene expression | EBV copies/106 T/B/NK cells |
|---|---|---|---|---|---|---|
| 1 | 1 | Bone marrow EBV+, CSF+; skin EBV+ HVLPD | T cell | 403 000 | Negative for EBNA1, BZLF1 | 23 400 T/ 5356 B |
| 2 | ND | Skin HVLPD, EBV ND | T∼B cell | 94 850 | ND | 249 T/ 451 B |
| 3 | 1 | Skin EBV+ HVLPD, bone marrow EBV+ | T cell | 4 510 000 | Negative for EBNA1, BZLF1 | 350 638 T/ 12 148 B |
| 4 | 1 | skin HVLPD, EBV ND | T cell | 583 500 | Negative for EBNA1, BZLF1 | 486 661 T/ 6966 B |
| 5 | ND | Skin HVLPD, EBV ND; ileum EBV+ suggestive of T-cell LPD | T cell | 196 500 | ND | ND |
| 6 | 1 | Skin EBV+ HVLPD | T cell | 728 500 | Positive for EBNA1, BZLF1 (3+), Neg for LMP1, EBNA2 | 250 616 T/ 20 000 B |
| 7 | 1 | EBV+ HVLPD | T cell | 250 150 | Positive for EBNA1; Negative for EBNA2, LMP1, BZLF1 | 199 050 T/ 1761 B |
| 8 | ND | ND | T>NK | 83 180* | ND | 227 227 T/313 B/13 514 NK |
| 9 | 1 | ND | NK∼T | 100 000* | ND | 93 343 T/958 B/113 907 NK |
| 10 | 1 | ND | NK>T | 245 500* | ND | 43 005 T/857 B/245 979 NK |
| 11 | 1 | Skin T cell EBV+ HVLPD, EBV+ lymph node | T cell | 320 000† | Positive for EBNA1, LMP1, BZLF1 (1+), Neg for EBNA2 | ND |
| 12 | 2 | Skin HVLPD, EBV ND | T cell | 840 350 | Positive for EBNA1, BZLF1 (3+); Negative for LMP1, EBNA2 | 452 353 T/227 778 B |
| 13 | 1 | Skin, colon, pleura, muscle EBV+ HVLPD | B>T cell | 1 514 800 | Negative for EBNA1, BZLF1 | 2 022 000 T/18 410 000 B (EBV was predominantly in T cells in the tissues) |
| 14 | 1 | Skin (EBV+ HVLPD), liver, bone marrow, lymph node | T>NK cell | 3 630 780* | Positive for EBNA1; Negative for BZLF1, EBNA2, LMP1 | 5 843 000 T/84 000 B/310 000 NK |
| 15 | 1 | Skin (EBV+ HVLPD), bone marrow, buccal and lingual mucosa,CSF with lymphocytic pleocytosis | T∼NK cell | 1 659 586* | Positive for EBNA1; Negative for BZLF1, EBNA2, LMP1 | 1 633 000 T/20 000 B/1 513 000 NK |
| 16 | ND | Skin EBV+ HVLPD, lung and ileum EBV+ T cell lymphoma | T>NK cell | 3 388 442* | ND | 872 000 T/3672 B/660 000 NK |
BZLF1 expression was graded 1+ to 4+.
CSF, cerebrospinal fluid; ND, not done.
IU/mL.
copies/106 cells.
A variety of treatments were tried (Table 1) for classic HVLPD, including hydroxychloroquine, valacyclovir, and topical and systemic corticosteroids; in general, only reduction of sun exposure was effective. For systemic HVLPD, while corticosteroids or thalidomide provided temporary improvements in signs and symptoms, only HSCT was curative.
Family histories were negative for photosensitivity or immunodeficiency. Targeted sequencing was performed for genes associated with immune deficiencies. Patient 13 had a mutation in GATA2, as reported previously12 ; none of the other patients had mutations in this gene. No mutations likely to result in loss of function were identified in genes associated with severe EBV disease16 or in genes associated with repair of DNA, which might be important for control of UV-induced DNA damage.17 No variants known to be pathogenic were found using whole-exome sequencing; analysis of these sequencing results is continuing, but results suggest that HVLPD is not a monogenic immunodeficiency.
T-cell subsets in patients with HVLPD
Only 2 patients had reduced numbers of CD4 and CD20 cells; T4/T8 ratios were reduced in both patients and an additional patient (Table 3). None of the patients had low numbers of CD8 cells or reduced levels of IgG. Ten of 16 patients had low NK-cell counts (5 whites and 5 nonwhites); 5 of 12 had elevated numbers of γ-δ T cells. Six of 13 patients had low numbers of effector memory CD4 cells; 2 of these 6 patients also had low numbers of effector memory CD8 cells (supplemental Table 1). Four of 13 patients had low numbers of central memory CD4 cells, while only 1 of these 4 patients also had low numbers of central memory CD8 cells. Three of 13 patients had low numbers of naive CD4 cells, and 2 of them also had low numbers of naive CD8 cells. Eight of 11 patients tested had elevated levels of Ki67+ CD3 cells in the blood indicative of proliferating T cells.
Hematologic features of HVLPD patients
| Patient | Race/sex | CD4 | CD8 | CD20 | NK | γ-δ T cells | Ki67/CD3 cells | T4/T8 ratio | IgG | Clonality |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | WM | 990 | 466 | 148 | 115 | 138 | 15 | 2.28 | 1038 | T-cell clone in blood and bone marrow |
| 2 | WF | 709 | 404 | 195* | 30 | ND | ND | ND | 864 | Polyclonal |
| 3 | WM | 619 | 652 | 367 | 357 | 939 | 44 | 1.28 | 827 | Indeterminate for T-cell clone in blood and bone marrow |
| 4 | WM | 466 | 304 | 334 | 564 | 109† | 10 | 1.53 | 866 | Indeterminate for T-cell clone |
| 5 | WM | 813 | 543 | 226* | 118 | ND | ND | ND | 1360 | T-cell clone in ileum and in blood |
| 6 | WF | 446 | 188 | 128 | 102 | 282 | 22 | 1.93 | 982 | Indeterminate for T-cell clone in blood |
| 7 | WF | 828 | 540 | 343 | 197 | 151 | 32 | 1.58 | 858 | Indeterminate for T-cell clone in blood; marrow polyclonal |
| 8 | WF | 676 | 289 | 220 | 120 | 131 | 32 | 2.34 | 1107 | Indeterminate for T-cell clone |
| 9 | WF | 1048 | 398 | 210 | 164 | 118 | 46 | 2.64 | 985 | Polyclonal |
| 10 | WF | 863 | 432 | 366 | 160 | 357 | ND | 2.00 | 910 | T-cell clone in blood |
| 11 | HM | 471 | 1166 | 160* | 77 | ND | ND | ND | 1910 | T-cell clone in skin, blood, and bone marrow |
| 12 | HM | 1395 | 462 | 186 | 117 | 111 | 18 | 3.06 | 1178 | T-cell clone in blood |
| 13 | AF | 122 | 1918 | 2 | 6 | ND | ND | 0.06 | 885 | T-cell clone in skin, blood, bone marrow, CSF, and BAL |
| 14 | AM | 203 | 1121 | 15 | 76 | 50 | 20 | 0.18 | 1760 | T-cell clone in blood |
| 15 | HF | 593 | 414 | 264 | 52 | 1326† | 114 | 1.43 | 1234 | Indeterminate for T-cell clone |
| 16 | HM | 443 | 890 | 341 | 211 | 1899 | 327 | 0.50 | 1557 | T-cell clone |
| Patient | Race/sex | CD4 | CD8 | CD20 | NK | γ-δ T cells | Ki67/CD3 cells | T4/T8 ratio | IgG | Clonality |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | WM | 990 | 466 | 148 | 115 | 138 | 15 | 2.28 | 1038 | T-cell clone in blood and bone marrow |
| 2 | WF | 709 | 404 | 195* | 30 | ND | ND | ND | 864 | Polyclonal |
| 3 | WM | 619 | 652 | 367 | 357 | 939 | 44 | 1.28 | 827 | Indeterminate for T-cell clone in blood and bone marrow |
| 4 | WM | 466 | 304 | 334 | 564 | 109† | 10 | 1.53 | 866 | Indeterminate for T-cell clone |
| 5 | WM | 813 | 543 | 226* | 118 | ND | ND | ND | 1360 | T-cell clone in ileum and in blood |
| 6 | WF | 446 | 188 | 128 | 102 | 282 | 22 | 1.93 | 982 | Indeterminate for T-cell clone in blood |
| 7 | WF | 828 | 540 | 343 | 197 | 151 | 32 | 1.58 | 858 | Indeterminate for T-cell clone in blood; marrow polyclonal |
| 8 | WF | 676 | 289 | 220 | 120 | 131 | 32 | 2.34 | 1107 | Indeterminate for T-cell clone |
| 9 | WF | 1048 | 398 | 210 | 164 | 118 | 46 | 2.64 | 985 | Polyclonal |
| 10 | WF | 863 | 432 | 366 | 160 | 357 | ND | 2.00 | 910 | T-cell clone in blood |
| 11 | HM | 471 | 1166 | 160* | 77 | ND | ND | ND | 1910 | T-cell clone in skin, blood, and bone marrow |
| 12 | HM | 1395 | 462 | 186 | 117 | 111 | 18 | 3.06 | 1178 | T-cell clone in blood |
| 13 | AF | 122 | 1918 | 2 | 6 | ND | ND | 0.06 | 885 | T-cell clone in skin, blood, bone marrow, CSF, and BAL |
| 14 | AM | 203 | 1121 | 15 | 76 | 50 | 20 | 0.18 | 1760 | T-cell clone in blood |
| 15 | HF | 593 | 414 | 264 | 52 | 1326† | 114 | 1.43 | 1234 | Indeterminate for T-cell clone |
| 16 | HM | 443 | 890 | 341 | 211 | 1899 | 327 | 0.50 | 1557 | T-cell clone |
Values in bold are below the limit of normal. Underlined values are above the upper limit of normal.
BAL, bronchoalveolar lavage; ND, not done.
CD19 cells measured.
γ-δ T cells were also present in the skin biopsy specimen. Normal values are as follows: CD4 cells, 359 to 1565; CD8 cells, 178 to 853; CD19 cells, 81 to 493; CD20 cells, 59 to 329; NK cells, 126 to 729, γ-δ T cells, 8 to 261, Ki67/CD3 cells, 9 to 18; T4/T8 ratio, 1.11 to 5.17; and IgG, 698 to 1560 mg/dL.
Patients with HVLPD had higher levels of CD8 T cells that recognize EBV latent membrane protein 2 (LMP2) compared with controls (P = .006; Figure 3A). No differences were seen in T cells that recognize an EBV lytic protein (BRLF1) compared with controls. Patients with HVLPD had reduced numbers of proliferating (CD127+) CD4 and CD8 T cells compared with healthy controls (P = .003 and 0.028, respectively; Figure 3B). Patients with HVLPD had lower levels of differentiated (CD27+), exhausted (PD1+), and homing (CCR7+) T cells that recognized EBV LMP2 compared with healthy controls (P < .001 [Figure 3C], P = .038 [Figure 3D], and P = .002 [Figure 3E], respectively).
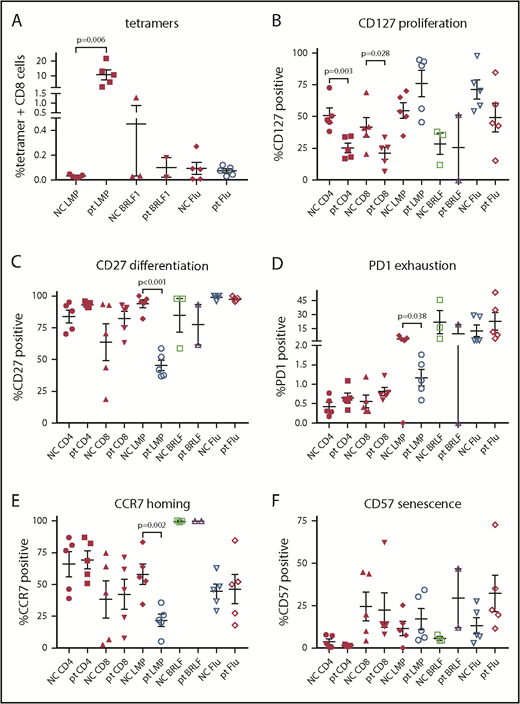
Lymphocyte subset and EBV tetramer staining of PBMCs from patients with HV and normal controls. Tetramer staining was performed for EBV LMP2, lytic protein BRLF1, and influenza M1 (flu) with costaining for CD8 (A), CD127 (B), CD27 (C), PD1 (D), CCR7 (E), or CD57 (F). P values are shown for results that are significantly different between patients and controls. NC, normal control; pt, patient.
Lymphocyte subset and EBV tetramer staining of PBMCs from patients with HV and normal controls. Tetramer staining was performed for EBV LMP2, lytic protein BRLF1, and influenza M1 (flu) with costaining for CD8 (A), CD127 (B), CD27 (C), PD1 (D), CCR7 (E), or CD57 (F). P values are shown for results that are significantly different between patients and controls. NC, normal control; pt, patient.
Antibody titers to EBV proteins
Most of our patients with HVLPD had EBV in their T cells like those with T-cell CAEBV without HVLPD, although HVLPD was often a more benign disease in white patients than T-cell CAEBV without HVLPD.11 Therefore, we compared EBV-specific antibody titers in patients with HVLPD (with either classic or systemic disease) and T-cell CAEBV without HVLPD as well as with healthy blood bank donors. Mean titers of antibodies to the EBV early protein BMRF1 (EBV DNA polymerase processivity factor) were significantly lower in patients with HVLPD than in those with T-cell CAEBV without HVLPD (P = .002) and similar to those in healthy EBV-seropositive blood bank donors (Figure 4). Mean titers of antibodies to EBV gp350 (major envelope glycoprotein), BHFR1 (bcl-2 homolog), BLRF2 (capsid protein), and BFRF3 (capsid protein) tended to be lower in patients with HVLPD than in those with T-cell CAEBV without HVLPD and more closely resembled titers observed in healthy blood bank donors. Mean antibody titers to EBV gH/gL (glycoproteins required for virus entry into cells) were higher in patients with HVLPD than in those with CAEBV without HVLPD and more closely resembled titers present in healthy blood-bank donors. Thus, patients with HVLPD tended to have titers of antibodies that more closely resembled healthy EBV-seropositive persons than those with T-cell CAEBV without HVLPD.

EBV protein antibody levels to gp350 (A), gH/gL (B), BHRF1 (C), BLRF2 (D), BFRF3 (E), and BMRF1 (F) in patients with HV, T-cell CAEBV without HVLPD, and healthy EBV-seropositive (EBV+) and EBV-seronegative (EBV−) blood-bank donors. Antibody levels were determined by immunoprecipitation and measured in light units.
EBV protein antibody levels to gp350 (A), gH/gL (B), BHRF1 (C), BLRF2 (D), BFRF3 (E), and BMRF1 (F) in patients with HV, T-cell CAEBV without HVLPD, and healthy EBV-seropositive (EBV+) and EBV-seronegative (EBV−) blood-bank donors. Antibody levels were determined by immunoprecipitation and measured in light units.
EBV DNA copy number, gene expression, and EBV typing in blood
Mean and median EBV copy number/mL of blood at the time of first evaluation at the NIH were 1 159 000 and 493 000 copies/mL, respectively (Table 2). Median viral loads at the time of first evaluation were higher (P = .013) in nonwhite patients (1 515 000 copies/mL) than in white patients (250 000 copies/mL). Similarly, median viral loads at the time of first presentation were higher in patients with systemic HVLPD (1 587 000 copies/mL) than classic HVLPD (327 000 copies/mL), but the difference was not significant (P = .098). At last follow-up, mean and median EBV copy numbers/mL of blood were 376 000 and 156 000 copies/mL in the 12 patients who did not undergo HSCT.
Examination of skin (Figure 2E-F) or blood showed that 11 out of 16 patients had EBV predominantly in T cells (7 white patients and 4 nonwhite patients), 2 had EBV at a similar level in T and NK cells, 1 had EBV predominantly in NK cells, 1 had EBV at a similar level in T and B cells, and 1 had EBV predominantly in B cells in the blood (although EBV was predominantly in T cells in the tissues) (Table 2). T-cell clones were found in the blood of 8 patients (5 of 6 nonwhite patients and 3 of 10 white patients), 6 had indeterminate or abnormal T-cell receptor patterns, and 2 were polyclonal. Five of 6 patients with systemic disease had a T-cell clone, while only 3 of 10 of those with classic HVLPD had a T-cell clone.
Most healthy EBV-seropositive adults have a type 0 pattern of EBV latency with no EBV latency genes expressed in peripheral blood cells.18 Type 1 latency indicates expression of EBNA1 alone, type 2 latency expression of EBNA1 and LMP1, and type 3 latency expression of EBNA1, EBNA2, and LMP1. Of the 10 patients tested, PBMCs from 4 patient showed type 0 latency, 5 had type 1 latency, and 1 had type 2 latency. Three of 10 patients showed EBV BZLF1 expression consistent with lytic EBV infection in PBMCs. Two types of EBV have been reported; all of the patients tested had type 1 EBV, except patient 12, who was born in South America.
Transcriptome of an HVLPD skin lesion
Comparison of the transcriptome of an EBV-positive HVLPD skin lesion with unaffected skin obtained the same day from patient 1 showed that genes encoding multiple chemokines were upregulated in the HVLPD skin biopsy specimen (Table 4). Among the genes most upregulated were CXCL11 and CXCL9, which encode chemoattractants for activated T cells; CXCL10, which encodes a chemoattractant for activated monocytes, T cells, and NK cells; and CCL4, which encodes a chemoattractant for monocytes and NK cells. Interestingly, CXCL11 (IFN-inducible T-cell alpha chemoattractant [I-TAC]), CXCL9 (monokine induced by interferon gamma [MIG]), and CXCL10 (IP-10) each interact with the chemokine receptor CXCR3, indicating the importance of this interaction in the pathogenesis of the skin lesions. Other upregulated genes included IFNG, which encodes interferon γ (IFN-γ) that inhibits EBV outgrowth,19 and APOBEC3A, which encodes a member of the cytidine deaminase family that inhibits replication of another human herpesvirus.20
Top 12 genes up- or downregulated in HVLPD skin compared with unaffected skin in patient 1 based on RNA-sequencing gene expression analysis
| Gene | Protein | Log2 fold increase or decrease (HVLPD vs unaffected skin) |
|---|---|---|
| Upregulated | ||
| CXCL11 | I-TAC | 15.91 |
| DCD | Dermcidin | 14.99 |
| SCGB2A2 | Secretoglobin | 14.82 |
| CXCL10 | IP-10 | 13.31 |
| IFNG | IFN-γ | 13.30 |
| SCGB1D2 | Secretoglobin family 1D member 2 | 13.05 |
| SERPINB4 | Serpin family B member 4 | 12.79 |
| PIP | Prolactin-induced protein | 12.75 |
| CXCL9 | MIG | 12.51 |
| IDO1 | Indoleamine 2,3 dioxygenase 1 | 12.21 |
| PRR4 | Proline-rich 4 | 11.76 |
| CCL4 | MIP1β | 11.71 |
| Downregulated | ||
| KRT79 | Keratin 79 | 11.84 |
| PM20D1 | Peptidase M20 domain containing 1 | 10.88 |
| AADACL3 | Arylacetamide deacetylase-like 3 | 10.76 |
| DGAT2L6 | Diacylglycerol O-acyltransferase 2 like 6 | 10.16 |
| AWAT2 | Acyl-CoA wax alcohol acyltransferase | 9.51 |
| SEC14L6 | SEC14-like lipid binding | 9.42 |
| FABP7 | Fatty acid binding protein 7 | 9.05 |
| CCL14 | CC-1 | 8.65 |
| THRSP | Thyroid hormone responsive | 8.59 |
| HAO2 | Hydroxyacid oxidase 2 | 8.46 |
| TMEM56 | Transmembrane protein 56 | 8.01 |
| UGT2A1 | UDP glucuronosyltransferase family 2 member A1 complex locus | 7.96 |
| Gene | Protein | Log2 fold increase or decrease (HVLPD vs unaffected skin) |
|---|---|---|
| Upregulated | ||
| CXCL11 | I-TAC | 15.91 |
| DCD | Dermcidin | 14.99 |
| SCGB2A2 | Secretoglobin | 14.82 |
| CXCL10 | IP-10 | 13.31 |
| IFNG | IFN-γ | 13.30 |
| SCGB1D2 | Secretoglobin family 1D member 2 | 13.05 |
| SERPINB4 | Serpin family B member 4 | 12.79 |
| PIP | Prolactin-induced protein | 12.75 |
| CXCL9 | MIG | 12.51 |
| IDO1 | Indoleamine 2,3 dioxygenase 1 | 12.21 |
| PRR4 | Proline-rich 4 | 11.76 |
| CCL4 | MIP1β | 11.71 |
| Downregulated | ||
| KRT79 | Keratin 79 | 11.84 |
| PM20D1 | Peptidase M20 domain containing 1 | 10.88 |
| AADACL3 | Arylacetamide deacetylase-like 3 | 10.76 |
| DGAT2L6 | Diacylglycerol O-acyltransferase 2 like 6 | 10.16 |
| AWAT2 | Acyl-CoA wax alcohol acyltransferase | 9.51 |
| SEC14L6 | SEC14-like lipid binding | 9.42 |
| FABP7 | Fatty acid binding protein 7 | 9.05 |
| CCL14 | CC-1 | 8.65 |
| THRSP | Thyroid hormone responsive | 8.59 |
| HAO2 | Hydroxyacid oxidase 2 | 8.46 |
| TMEM56 | Transmembrane protein 56 | 8.01 |
| UGT2A1 | UDP glucuronosyltransferase family 2 member A1 complex locus | 7.96 |
CoA, coenzyme A; UDP, uridine diphosphate.
Among the 10 genes most downregulated in the HVLPD lesion were genes that encode proteins that are highly expressed in the skin (KRT79 and AADACL3) or genes that encode proteins important for lipid and fatty acid metabolism (PM20D1, AWAT2, SEC14L6, and FABP7).
Cytokine levels in patients with EBV HV
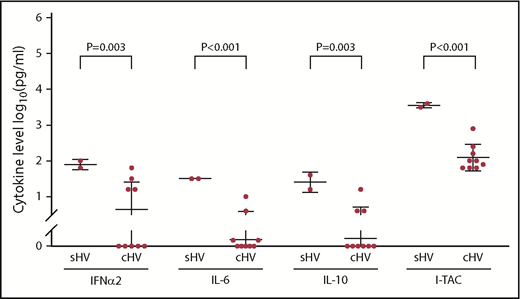
The mean serum levels of the cytokines tested in 11 patients with EBV HVLPD were similar to 13 healthy controls (all P values > .01). In contrast, the mean level of all but one of the cytokines tested in 12 patients with EBV HVLPD was lower than those in 10 patients with T-cell CAEBV without HVLPD; serum levels of 6 cytokines (interleukin-6 [IL-6], IL-18, tumor necrosis factor α [TNF-α], IP-10, MIG, and I-TAC) were significantly lower in patients with EBV HVLPD than in T-cell CAEBV patients without HVLPD (P = .003, P < .01, P = .002, P = .004, P < .001, and P < .001, respectively) (Figure 5). Serum levels of 6 cytokines (IL-8, IL-18, TNF-α, IP-10, MIG, and I-TAC) were all significantly higher in T-cell CAEBV patients without HVLPD than healthy controls (P = .002, P = .002, P = .003, P < .001, P < .001, and P < .001, respectively). Thus, the level of serum cytokines in patients with EBV HVLPD resembled that of normal controls more than patients with T-cell CAEBV without HVLPD.

Serum cytokine levels in patients with HV, T-cell CAEBV without HVLPD, and EBV-seropositive blood-bank donor controls (CTRL).
Serum cytokine levels in patients with HV, T-cell CAEBV without HVLPD, and EBV-seropositive blood-bank donor controls (CTRL).
Mean serum levels of most cytokines (IL-6, IL-10, IL-18, IP10, MCP-1, TNF-α, MIG, I-TAC, IFN-α2, and IFN-γ) were higher in Asian and Hispanic patients than in whites, but the differences were not significant. Significantly higher levels of IFN-α2, IL-6, IL-10, and I-TAC were noted in patients with systemic disease than in those with classic HVLPD (P = .003, P < .001, P = .003, and P < .001, respectively) (Figure 6).
Follow-up
Four nonwhite patients (patients 11, 13, 14, and 16) underwent HSCT due to systemic disease and persistent fever, T-cell lymphoma, or hepatitis. After transplant, EBV levels were undetectable in 3 patients and in the low-thousand range of copies/mL for the fourth patient. Patients 11 and 13 are doing well after HSCT; however, patient 14 developed an EBV-positive nasal NK/T-cell lymphoma after having undergone a haploidentical transplant; he is currently in remission after receiving chemotherapy and radiation therapy. Patient 16 has idiopathic pulmonary syndrome now 1 month after transplant. Patient 15 has central nervous system disease and is being evaluated for HSCT.
One of the white patients (patient 5) developed systemic disease with lymphoproliferative disease involving the gastrointestinal tract at age 23 years, which resolved without therapy. Two whites with classic HVLPD (now ages 12 and 17 years) have had no new HVLPD skin lesions despite unprotected sun exposure (patients 4 and 7); both have had a drop in the level of EBV DNA in the blood from ∼250 000 copies/mL to 10 000 to 30 000 copies/mL.
The mean duration since onset of disease for all 16 patients (both white and nonwhite) was 12 years (range, 7-22 years). The total duration of follow-up since onset of disease for the entire cohort is 190 patient-years.
Discussion
We reviewed our 11-year experience with EBV HVLPD at NIH and found that patients in the United States and Great Britain exhibit several differences when compared with the largest series of patients with HVLPD to date reported by Hirai and colleagues in Japan.4 Approximately 60% (10/16) of patients in our study were white, while 100% of 54 patients (33 classic, 15 systemic, and 6 with HVLPD and HMB) reported in the series from Japan were Asian. None of our patients had HVLPD with HMB, while 11% from Japan had this symptom. Approximately 60% (10/16) of our patients and those from Japan (33/54) had classic disease without systemic symptoms. Only 10% (1/10) of our white patients had systemic disease that resolved without therapy, while 83% (5/6) of our Asian and Hispanic patients had systemic disease (P = .008). Reduced numbers of NK cells were present in 63% (10/16) of our patients, a finding that was not reported in the patients from Japan. At present, after 190 patient-years of follow-up, none of our patients have died of their disease, although 4 patients with systemic disease required HSCT. In the series of Japanese patients, 3 patients with systemic HVLPD and HMB died of progressive disease or hemophagocytic syndrome. Thus, our patients tended to have less severe disease than those from Japan.
In our study, nonwhites had a later onset of HVLPD than whites (median age, 8 vs 5 years), higher EBV DNA levels in the blood (median, 1 515 000 vs 250 000 copies/mL), and higher levels of cytokines in the serum and were more likely to have a T-cell clone in the blood (83% vs 30% of patients). The mean duration of follow-up was similar for nonwhite and white patients (11 years). Few case series have reported white patients with HV. In two of these studies,21,22 EBV levels in the blood or tissues were not examined, none of the patients developed systemic disease, and 22%21 to 60%22 had spontaneous resolution of their disease. In a more recent study of 7 patients from France, all of whom had elevated EBV DNA in the blood, patients were followed from 4 to 39 years after their initial skin lesions, and none developed systemic disease; 3 had spontaneous remissions.23 Only 1 of our patients had EBV predominantly in NK cells; in contrast, studies from Mexico8 and Japan5 indicate that 30% to 40% of patients have EBV in NK cells.
Many of the differences observed in nonwhite compared with white patients were also seen in persons with systemic vs classic HVLPD. Patients with systemic HVLPD had higher median EBV DNA levels in the blood than those with classic HVLPD (1 587 000 vs 327 000 copies/mL), were more likely to have a T-cell clone in blood (83% vs 30%), and had higher levels of serum cytokines. Although some studies have suggested that persons with HVLPD and higher viral loads are more likely to have a more severe form of the disease,2 a more recent study showed no correlation between the level of EBV DNA and poor prognosis.6
We observed several abnormalities in lymphocyte populations that may be important for control of EBV. Approximately 60% of patients had reduced numbers of NK cells, which are critical for controlling EBV disease.24 Approximately 75% of our patients had increased numbers of Ki67-positive T cells, indicative of proliferating cells. Approximately 40% of our patients had elevated numbers of γ-δ T cells; in contrast, γ-δ T cells have been reported in up to 90% of patients from Asia.4 Approximately 40% of patients had low numbers of effector memory CD4 cells, and one-third of these patients also had low numbers of effector memory CD8 cells. Effector memory cells produce cytokines such as IFN-γ in response to antigen stimulation and can rapidly respond to antigens, but their expansion is limited, which can prevent eradication of pathogens. Approximately 25% of patients had low numbers of naive CD4 T cells, and two-thirds of these patients also had low numbers of naive CD8 cells. These cells have not yet encountered foreign antigens, and they are important for the immune response to novel pathogens. Reduced numbers of effector memory and naive CD4 and CD8 cells were also often observed in patients with T-cell CAEBV without HVLPD (J.I.C., unpublished data, 19 December 2018). In contrast, increased numbers of effector memory T cells are present in healthy persons during primary EBV infection.25 Patients with HVLPD had higher levels of T cells that recognize EBV LMP2, and these cells showed higher levels of proliferation and an effector memory phenotype compared with healthy controls. The persistently high level of EBV in the blood of these patients suggests that these T cells may have impaired functional activity.
Most of our patients had EBV infection predominantly in their T cells. Coleman et al showed that EBV type 2 has a predilection for infecting T cells in vitro15 and in vivo26 compared with EBV type 1, which infects B cells. In contrast, all but one of our patients were infected with type 1 EBV. Thus, type 1 is fully capable of infecting T cells in vivo. Infection of T cells in vitro with EBV results in dysregulation of cytokine production with high levels of MIP-1α, MIP-1β, and IL-8.15 However, serum levels of these cytokines in our patients with EBV predominantly in circulating T cells were not elevated. Thus, in vitro infection of T cells with EBV does not recapitulate the cytokine patterns seen with T-cell infection in patients with HV.
In the one patient for whom we could obtain HVLPD and normal tissue on the same day, we found multiple chemokine genes upregulated in HVLPD tissue compared with unaffected tissue, including genes encoding I-TAC, IP-10, MIG, and MIP-1α. Oshima et al reported upregulation of genes encoding I-TAC, IP-10, MIG, and MIP-1α in tissues from patients with EBV-positive NK-cell lymphoma but downregulation of genes encoding I-TAC, IP10, and MIG in tissues from patients with CAEBV compared with tissues with nonspecific lymphadenitis.27 Teruya-Feldstein et al showed that genes encoding MIG and IP-10 were increased in tissues from patients with EBV-positive T/NK-cell lymphomas compared with controls with lymphoid hyperplasia.28 Setsuda et al found increased levels of genes encoding MIG, IP-10, and MIP-1α in patients with EBV-positive infectious mononucleosis compared with those from patients with EBV posttransplant lymphoproliferative disease.29 Thus, the pattern of cytokine genes upregulated in HVLPD tissue resembled that seen in patients with EBV-positive NK-cell lymphoma and infectious mononucleosis but differed from that seen in patients with CAEBV.
We did not find germline mutations in genes previously associated with severe EBV disease or UV damage. It is possible that HVLPD may be associated not with germline mutations but instead with somatic mutations in lymphocytes, as has been seen in CAEBV.30
HVLPD has many similarities to CAEBV without HVLPD, including high levels of EBV DNA in peripheral blood, EBV present predominantly in T or NK cells, a high frequency of clonal T cells in the blood, and risk for progression to T- or NK-cell lymphoma.2-5,11,31 In our prior study of patients with CAEBV without HVLPD, all of whom died in the absence of transplant,11 the median EBV load was 320 000 copies per million cells. Here, we found that patients with HVLPD had a median EBV load of 493 000 copies/mL of blood. Thus, while patients with either HVLPD or CAEBV without HVLPD have very high levels of EBV predominantly in non-B cells, the prognosis for these patients, particularly whites, can be very different. Our previous study showed that patients with severe CAEBV without HPLVD had significantly elevated serum levels of several cytokines, including IL-6, IFN-γ, and TNF-α, compared with healthy controls11 ; here we found that the levels of these 3 cytokines in patients with HVLPD were comparable to levels in healthy controls and lower than those in patients with T-cell CAEBV without HVLPD. Thus, difference in serum cytokines might be related to the difference in pathogenesis and prognosis of HVLPD and CAEBV without HVLPD.
Limitations to our study of this rare disease include the modest numbers of patients, limited number of biopsy specimens, and the narrow age range (all but one of our patients were 11-27 years old at the last follow-up).
In summary, we found that white patients with HVLPD have a better prognosis, less systemic disease, lower EBV DNA copy numbers in the blood, less likelihood of having T-cell clones, and higher NK-cell levels than nonwhite patients. Other EBV-associated diseases, such as nasopharyngeal carcinoma or CAEBV, are more common in certain populations, including Asians, than in Europeans and North Americans, suggesting that genetic or environmental differences may account for the increased severity of certain EBV diseases. HVLPD also shares many features in common with CAEBV without HVLPD, including high levels of EBV DNA in the blood primarily in T or NK cells and a high frequency of clonal T cells. However, while CAEBV without HVLPD is invariably fatal in the absence of HSCT, many patients with HV, particularly whites, have a good prognosis and may not require HSCT. Thus, it is important to distinguish HVLPD from CAEBV without HVLPD and to follow HVLPD patients who have no systemic symptoms conservatively.
Qualified researchers may request access to data by contacting the corresponding author (jcohen@niaid.nih.gov).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Gillian Hooker for help with genetic counseling and Jing Qin for help with statistics. Tetramers for EBV LMP2, EBV BRLF1, and influenza M1 were obtained through the NIH Tetramer Core Facility.
This work was supported by the intramural research programs of the National Institutes of Health (NIH), National Institute of Allergy and Infectious Diseases, the NIH, National Cancer Institute, and the NIH, National Human Genome Research Institute. This project has been funded in part with federal funds from the NIH, National Cancer Institute, under contract number HHSN261200800001E.
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Authorship
Contribution: J.I.C. and I.M. designed the study; J.I.C. supervised the study and wrote the first draft of the manuscript; I.M., K.H.K., J.J.D., S.P., and E.S.J. helped with editing the manuscript; I.M., H.F., R.P.S., J.J.D., and K.H.K. provided data on patients and participated in clinical care and decision-making on patients; K.D., T.A.K., P.R., R.L.H., and W.B. performed laboratory assays; L.L.B., P.S.C., A.J.S., and F.S.C. performed genetic testing and analysis; S.-P.T., M.A.L., D.T., and K.L. assisted with patient care; T.G.M. performed RNA-sequencing analysis; E.S.J. and S.P. were responsible for diagnosis and reviewing pathology; and all authors reviewed the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Peter S. Chines died on 10 July 2017.
Correspondence: Jeffrey I. Cohen, Laboratory of Infectious Diseases, Building 50, Room 6134, National Institutes of Health, 50 South Dr, MSC8007, Bethesda, MD 20892; e-mail: jcohen@niaid.nih.gov.
