

Key Points
SCD alters the composition of the clot in a mouse model of venous thrombosis.
Therapeutic RBC exchange partially normalizes retraction and resistance to fibrinolysis of whole blood sickle clots.

Abstract
Sickle cell disease (SCD) is associated with chronic activation of coagulation and an increased risk of venous thromboembolism. Erythrocyte sickling, the primary pathologic event in SCD, results in dramatic morphological changes in red blood cells (RBCs) because of polymerization of the abnormal hemoglobin. We used a mouse model of SCD and blood samples from sickle patients to determine if these changes affect the structure, properties, and dynamics of sickle clot formation. Sickling of RBCs and a significant increase in fibrin deposition were observed in venous thrombi formed in sickle mice. During ex vivo clot contraction, the number of RBCs extruded from sickle whole blood clots was significantly reduced compared with the number released from sickle cell trait and nonsickle clots in both mice and humans. Entrapment of sickled RBCs was largely factor XIIIa–independent and entirely mediated by the platelet-free cellular fraction of sickle blood. Inhibition of phosphatidylserine, but not administration of antisickling compounds, increased the number of RBCs released from sickle clots. Interestingly, whole blood, but not plasma clots from SCD patients, was more resistant to fibrinolysis, indicating that the cellular fraction of blood mediates resistance to tissue plasminogen activator. Sickle trait whole blood clots demonstrated an intermediate phenotype in response to tissue plasminogen activator. RBC exchange in SCD patients had a long-lasting effect on normalizing whole blood clot contraction. Furthermore, RBC exchange transiently reversed resistance of whole blood sickle clots to fibrinolysis, in part by decreasing platelet-derived PAI-1. These properties of sickle clots may explain the increased risk of venous thromboembolism observed in SCD.
Introduction
Chronic activation of coagulation is commonly observed in sickle cell patients and in mouse models of sickle cell disease (SCD).1,2 Studies from our group and others have demonstrated that the hypercoagulable state contributes to chronic vascular inflammation and end organ damage in mouse models of SCD.3-6 Furthermore, SCD is characterized by an increased risk of thrombotic complications.7-11 The presence of “in situ” thrombi in the pulmonary vasculature and brain has been reported at autopsy in SCD patients,12-15 and case studies have demonstrated thrombosis within the hepatic vein and inferior vena cava (IVC).16,17 Epidemiologic studies have reported an increased incidence of venous thromboembolism (VTE) in SCD patients, even after accounting for frequency of hospitalization.11 Importantly, VTE in patients with SCD is associated with a high recurrence rate and greater mortality.8,11
Recent studies strongly implicate the contribution of red blood cells (RBCs) to both hemostasis and thrombosis, including their key role in VTE and in the clot contraction process.18-23 RBCs compose a significant part of thrombi, especially those formed in veins (so called “red thrombi”).24 Fibrin and platelets form a network on the exterior of the clot, whereas biconcave RBCs are compacted and compressed internally, adopting polyhedral shapes.21 Clot contraction is mediated by contractile forces applied by activated platelets on the fibrin network, which over time increases clot density, improves its stability, and decreases clot size.21,22,25,26 In normal whole blood, retention of RBCs within the contracting clot depends on fibrin α-chain crosslinking by factor XIIIa.19,27
Sickling of RBCs is the primary pathologic event in SCD. Under hypoxic conditions, sickle RBCs undergo a dramatic change in morphology resulting from polymerization of abnormal hemoglobin (Hb) tetramers.28 These changes result in formation of sickle-shaped RBCs with reduced flexibility and the extrusion of multiple spiculelike processes.29 Because of these changes, it would be expected that SCD might affect the properties of the clot. For example, decreased RBC deformability reduces fibrin network permeability, which may render clots more resistant to fibrinolytic agents.30 In addition, artificial incorporation of sickle RBCs into ex vivo–formed fibrin clot results in more heterogeneous and agglomerated fibrin matrix structures compared with those formed with healthy RBCs.31 Finally, it has been reported that packaging of RBCs during ex vivo clot contraction is altered in the blood of SCD patients.22,32
In this study, we used a mouse model of SCD and blood samples from sickle patients to determine if SCD affects the dynamics of venous thrombosis in vivo, and the properties of mouse and human clots formed ex vivo. We also investigated effects of therapeutic RBC exchange on the properties of clots in patients with SCD.
Materials and methods
Detailed information regarding experimental design is available in the supplemental Materials and methods, available on the Blood Web site.
Human subjects
Blood was obtained from consenting subjects in accordance with the Declaration of Helsinki and the University of North Carolina and University of Lyon Institutional Review Boards.
Mice
Generation of nonsickle (AA) and sickle cell (SS) Townes mice that have both human α- and β- (βA and βS form) globin genes knocked into the mouse locus have been previously described.33 All animal studies were approved by the University of North Carolina Animal Care and Use Committees in compliance with National Institutes of Health guidelines.
Statistical analysis
Statistical analysis was performed using GraphPad Prism software (version 6.07; GraphPad Software, Inc., La Jolla, CA). The significance level was defined as P < .05; all data are displayed as means ± standard error of the mean.
Results
Sickling of RBCs coincides with enhanced venous thrombosis in sickle mice
To investigate if SCD affects properties and dynamics of venous thrombus formation in vivo, we initially attempted to use a well-accepted mouse model of venous thrombosis induced by IVC stenosis.34 Unfortunately, all SS mice (n = 10) subjected to this model died shortly after initiating the surgical procedure. We were only able to obtain a clot from the single SS mouse that survived 2 hours after surgery. Thrombi collected from nonsickle (AA) mice 2 hours after surgery contained regular-shaped RBCs that were densely packed within the fibrin network (Figure 1A). Interestingly, and in contrast to the control thrombi, the thrombus from the sickle mouse contained RBCs that had undergone extensive sickling, characterized by a dramatic shape change and formation of long, spiculelike processes (Figure 1B). In addition, we observed a striking increase in the deposition of acellular material within the sickle thrombus (Figure 1C), which on high-magnification images looked like a dense fibrin network (Figure 1D).

Sickling of RBC and increased deposition of acellular material within venous thrombi formed in SCD mice. Scanning electron microscopy of clots formed 2 hours after inferior vena cava stenosis in AA (A) and SS mice (B-D). Original magnification: ×8000 (A), ×5000 (B-C), and ×20 000 (D).
Sickling of RBC and increased deposition of acellular material within venous thrombi formed in SCD mice. Scanning electron microscopy of clots formed 2 hours after inferior vena cava stenosis in AA (A) and SS mice (B-D). Original magnification: ×8000 (A), ×5000 (B-C), and ×20 000 (D).
Because of the high mortality rate in SS mice subjected to the IVC stenosis model, we proceeded to use a less invasive model, namely electrolytic injury–induced thrombosis in the femoral vein.35,36 This model allows real-time observation of fibrin and platelet deposition within developing, nonocclusive thrombi (Figure 2A). In AA mice, platelet accumulation reached a plateau within 15 minutes after injury and was sustained throughout the monitoring period (Figure 2B). A modest increase in platelet accumulation observed in SS and heterozygous AS mice (a model of sickle cell trait) was not statistically different from that observed in AA mice (Figure 2B; supplemental Videos 2-4). In contrast, fibrin accumulation in SS thrombi was significantly increased compared with fibrin deposition in thrombi from AA mice (Figure 2C; supplemental Videos 2 and 4), despite similar levels of circulating fibrinogen (supplemental Figure 2). Interestingly, AS mice demonstrated a pattern of fibrin deposition that was intermediate between AA and SS mice, although this difference was not statistically significant (Figure 2C; supplemental Video 3).
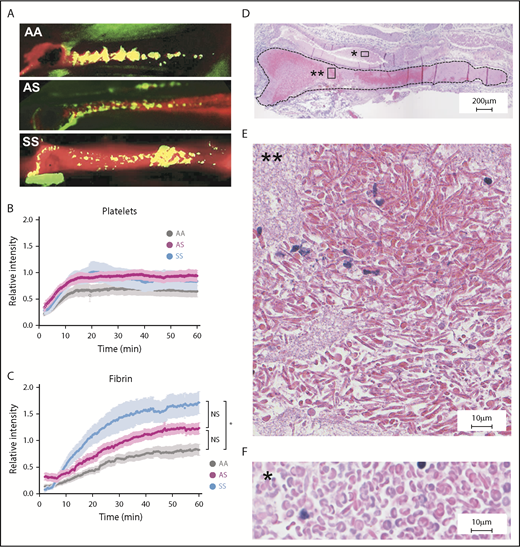
Increased fibrin deposition and evidence of RBC sickling in thrombi formed in the femoral vein of SCD mice. (A) Representative images of platelet (green) and fibrin (red) deposition within thrombi formed in the femoral vein of AA, AS, and SS mice 60 minutes after electrolytic injury. Yellow color shows overlap between fibrin and platelets. Representative movies demonstrating the dynamics of clot formation over 1 hour are included as supplemental Videos 2-4 for AA, AS, and SS mice, respectively. Intravital microscopy analysis of platelet accumulation (B) and fibrin deposition (C) within thrombi of AA (n = 14, gray line), AS (n = 9, purple line), and SS (n = 10, blue line) mice quantitated as a relative intensity. *P < .05. (D) Hematoxylin and eosin staining of the thrombus (delineated by black dotted line) formed in the femoral vein of SS mice demonstrates the presence of RBCs (dark pink) and fibrin/platelets (light pink). Extensive sickling of the RBCs is present within the thrombus (E) but not in the blood pool outside the thrombus (F). Original magnification: ×4 (D) and ×40 (E-F). NS, not significant.
Increased fibrin deposition and evidence of RBC sickling in thrombi formed in the femoral vein of SCD mice. (A) Representative images of platelet (green) and fibrin (red) deposition within thrombi formed in the femoral vein of AA, AS, and SS mice 60 minutes after electrolytic injury. Yellow color shows overlap between fibrin and platelets. Representative movies demonstrating the dynamics of clot formation over 1 hour are included as supplemental Videos 2-4 for AA, AS, and SS mice, respectively. Intravital microscopy analysis of platelet accumulation (B) and fibrin deposition (C) within thrombi of AA (n = 14, gray line), AS (n = 9, purple line), and SS (n = 10, blue line) mice quantitated as a relative intensity. *P < .05. (D) Hematoxylin and eosin staining of the thrombus (delineated by black dotted line) formed in the femoral vein of SS mice demonstrates the presence of RBCs (dark pink) and fibrin/platelets (light pink). Extensive sickling of the RBCs is present within the thrombus (E) but not in the blood pool outside the thrombus (F). Original magnification: ×4 (D) and ×40 (E-F). NS, not significant.
The small size of the clots generated in this model prevented a reliable analysis of the clot weight. However, histological analysis revealed presence of RBCs in both AA (data not shown) and SS clots (Figure 2D). Importantly, RBCs incorporated within thrombi formed in SS mice underwent extensive sickling (Figure 2E), consistent with that seen in the thrombus formed in the IVC stenosis model. Of note, although RBC sickling was observed within the thrombi, RBCs outside the thrombi exhibited normal morphology (Figure 2F).
SCD reduces RBC extrusion during clot contraction
During clot contraction, RBCs may be extruded into the surrounding milieu.21,22 We therefore investigated whether RBC sickling observed within the venous thrombi affects the RBC retention in contracting clots. We used a previously described ex vivo assay to quantify the number of RBCs extruded from the clot.19,20 The number of murine SS RBCs extruded into the serum during clot contraction was dramatically reduced compared with the number of AA or AS RBCs (Figure 3A-B). We have previously shown that factor XIII (FXIII) activity promotes retention of RBCs within contracting clots and thereby enhances thrombus size.19 As expected, inhibition of FXIIIa activity with T101 (10 µM) increased release of AA RBCs from murine whole blood clots (Figure 3A). A similar effect of T101 was observed for AS RBCs. However, in contrast, inhibition of FXIIIa activity did not promote extrusion of SS RBCs (Figure 3A).

Inhibition of clot contraction-mediated extrusion of mouse sickle RBCs from whole blood clots formed ex vivo. (A) Number of RBCs released from clots formed ex vivo from the blood of AA (n = 15), AS (n = 10), and SS (n = 16) mice in the absence (−) or presence (+) of FXIIIa inhibitor T101 (20 µM). ***P < .001. (B) Representative images of whole blood (before initiation of clot formation) and serum (after removal of clots formed for 2 hours). Yellow color of SS serum indicates sparse RBC presence. (C) Effect of changes in hematocrit on the serum RBC content 2 hours after contraction of the clot formed from the blood of AA (n = 9) and SS (n = 11) mice. (D) Scanning electron micrographs of ex vivo whole blood clots. Arrows indicate fibrin, arrowheads indicate sickled RBC processes. Original magnification: ×10 000 (upper) and ×40 000 (lower). (E) Transmission electron micrographs of clots formed ex vivo from the blood of AA and SS mice. Original magnification ×10 000. Gen, genotype.
Inhibition of clot contraction-mediated extrusion of mouse sickle RBCs from whole blood clots formed ex vivo. (A) Number of RBCs released from clots formed ex vivo from the blood of AA (n = 15), AS (n = 10), and SS (n = 16) mice in the absence (−) or presence (+) of FXIIIa inhibitor T101 (20 µM). ***P < .001. (B) Representative images of whole blood (before initiation of clot formation) and serum (after removal of clots formed for 2 hours). Yellow color of SS serum indicates sparse RBC presence. (C) Effect of changes in hematocrit on the serum RBC content 2 hours after contraction of the clot formed from the blood of AA (n = 9) and SS (n = 11) mice. (D) Scanning electron micrographs of ex vivo whole blood clots. Arrows indicate fibrin, arrowheads indicate sickled RBC processes. Original magnification: ×10 000 (upper) and ×40 000 (lower). (E) Transmission electron micrographs of clots formed ex vivo from the blood of AA and SS mice. Original magnification ×10 000. Gen, genotype.
Because SCD is associated with a reduced hematocrit, we next investigated if the initial RBC number affects the extrusion of these cells during clot contraction. Indeed, lowering hematocrit in AA mouse blood reduced RBC extrusion from the clots (Figure 3C). However, increasing the hematocrit in SS mouse blood to that of AA blood did not result in a proportional increase of extruded SS RBCs (Figure 3C). In aggregate, these data indicate that reduced extrusion of RBCs from SS clots does not simply reflect lower RBC numbers and is not affected by blocking FXIIIa activity.
Entrapment of mouse sickle RBCs within the clot is not mediated by platelets or plasma components of sickle blood
To identify the mechanism responsible for sickle RBC entrapment within the clot, we characterized the structure of sickle clots formed ex vivo. Scanning electron microscopy demonstrated that RBCs within AA clots had polyhedral shapes and were tightly packed in the central part of the clot (Figure 3D, left). In contrast, most of the mouse SS RBCs were not compacted within the clot and had heterogeneous shapes caused by Hb sickling within these cells (Figure 3D, right). The most striking feature of the sickle clots was the presence of long membrane extensions originating from the sickled RBCs (supplemental Figure 3). These spiculelike processes intertwined with each other and with the fibrin network (Figure 3D, lower right). The profound differences in the cellular organization of AA and SS clots were also apparent by transmission electron microscopy (Figure 3E). These data suggest that the entrapment of sickle RBCs within the clot may be mediated, in part, by the structural properties of these cells. To test this hypothesis, we investigated which compartment of sickle blood, platelet-poor plasma (PPP) or cellular fraction, mediates the difference in RBC extrusion from whole blood clots during clot contraction. Experimental procedures involved in preparation and mixing PPP and the cellular fraction of AA or SS mouse blood (Figure 4A-B) did not affect the initial number of RBCs and platelets observed in the unaltered blood of AA or SS mice (data not shown). Importantly, this experiment demonstrated that entrapment of SS RBCs is primarily mediated by the cellular fraction of sickle blood (Figure 4C) and occurred despite an elevated number of platelets in SS blood (Figure 4B). To further identify the cell type responsible for this difference, we mixed PPP, platelets, and the platelet-free cellular fraction of AA or SS mouse blood. As shown in Figure 4D, entrapment of SS RBCs within the clot was entirely mediated by the platelet-free cellular fraction of sickle blood containing RBCs.
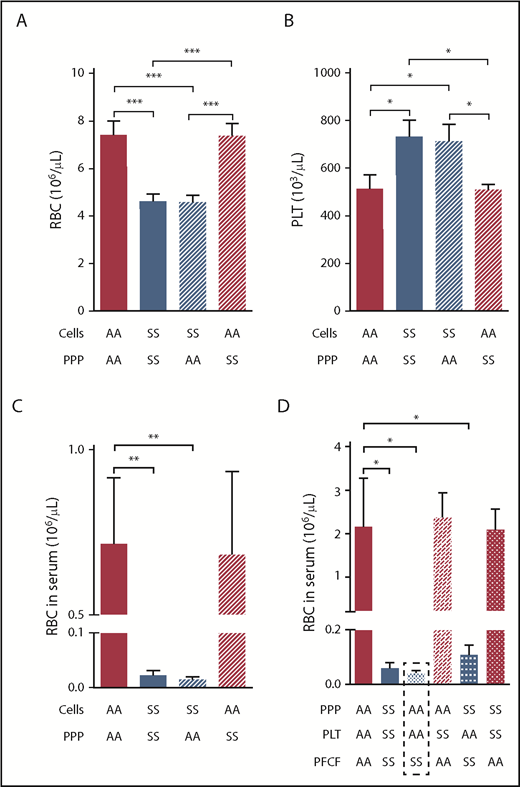
Entrapment of RBCs within the mouse sickle clot is mediated by the platelet-free cellular blood compartment. Initial RBC (A) and platelet number (B) after recombining PPP and cellular compartments of AA or SS mouse blood at indicated combinations followed by analysis of RBC number released from the clots into the serum (C; n = 4 per group). (D) Serum RBC content after clot contraction performed with reconstituted whole blood samples from different combinations of PPP, PLT, and PFCF of the blood from AA or SS mice (n = 6 per group). *P < .05, **P < .01, ***P < .001. PFCF, platelet-free cellular fraction; PLT, platelets.
Entrapment of RBCs within the mouse sickle clot is mediated by the platelet-free cellular blood compartment. Initial RBC (A) and platelet number (B) after recombining PPP and cellular compartments of AA or SS mouse blood at indicated combinations followed by analysis of RBC number released from the clots into the serum (C; n = 4 per group). (D) Serum RBC content after clot contraction performed with reconstituted whole blood samples from different combinations of PPP, PLT, and PFCF of the blood from AA or SS mice (n = 6 per group). *P < .05, **P < .01, ***P < .001. PFCF, platelet-free cellular fraction; PLT, platelets.
SCD affects ex vivo clot retraction in human blood
Consistent with our observations in mouse blood, extrusion of RBCs from clots was also reduced during contraction of whole blood clots from sickle cell patients (SS), compared with both sickle trait (AS) and healthy (AA) subjects (Figure 5A). Inhibition of FXIIIa activity significantly increased the number of RBCs extruded from AA and AS clots (Figure 5A). Interestingly however, this increase was significantly higher in AA than AS clots. A moderate, twofold increase in the number of released RBCs observed for SS clots after FXIIIa inhibition did not reach statistical significance (Figure 5A).
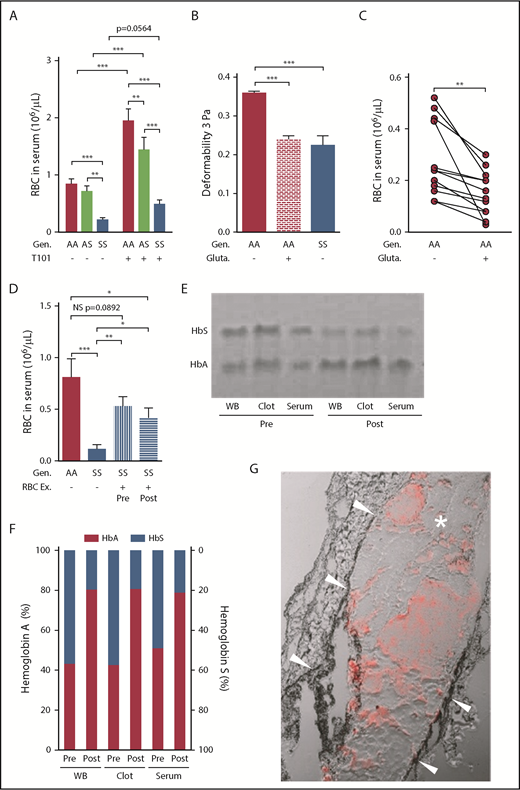
RBC retention in sickle whole blood clots is mediated, in part, by reduced RBC deformability, and can be partially reversed by FXIIIa inhibition or RBC exchange. (A) Number of RBCs released from clots formed ex vivo from the blood of AA subjects (n = 20), AS (n = 13), and SS patients (n = 29) in the absence (−) or presence (+) of FXIIIa inhibitor T101 (20 µM). (B) Effects of glutaraldehyde (0.1%) on AA RBC (n = 6) deformability compared with the deformability of AA (n = 6) and SS (n = 5) RBCs incubated with cold isotonic buffer at 3 Pa. (C) Serum RBC content after clot contraction performed with PRP reconstituted with AA RBCs exposed to isotonic buffer or 0.1% glutaraldehyde (n = 13). (D) Number of RBCs released from clots formed ex vivo from blood obtained from AA control subjects (n = 8), SS patients (n = 11), or SS patients undergoing RBC Ex pre- and postprocedure (n = 10). Representative gel (E) and analysis of hemoglobin content (F) in the WB, clots, and serum obtained from pre- and postexchange samples (n = 5). (G) Representative image of the clot formed in the femoral vein of SS mice (n = 4) injected with AA RBCs. Arrowheads point to the vessel wall; asterisk indicates the clot; red staining demonstrates the presence of AA RBCs within the clot. *P < .05, **P < .01, ***P < .001. Ex, exchange; gluta, glutaraldehyde; WB, whole blood.
RBC retention in sickle whole blood clots is mediated, in part, by reduced RBC deformability, and can be partially reversed by FXIIIa inhibition or RBC exchange. (A) Number of RBCs released from clots formed ex vivo from the blood of AA subjects (n = 20), AS (n = 13), and SS patients (n = 29) in the absence (−) or presence (+) of FXIIIa inhibitor T101 (20 µM). (B) Effects of glutaraldehyde (0.1%) on AA RBC (n = 6) deformability compared with the deformability of AA (n = 6) and SS (n = 5) RBCs incubated with cold isotonic buffer at 3 Pa. (C) Serum RBC content after clot contraction performed with PRP reconstituted with AA RBCs exposed to isotonic buffer or 0.1% glutaraldehyde (n = 13). (D) Number of RBCs released from clots formed ex vivo from blood obtained from AA control subjects (n = 8), SS patients (n = 11), or SS patients undergoing RBC Ex pre- and postprocedure (n = 10). Representative gel (E) and analysis of hemoglobin content (F) in the WB, clots, and serum obtained from pre- and postexchange samples (n = 5). (G) Representative image of the clot formed in the femoral vein of SS mice (n = 4) injected with AA RBCs. Arrowheads point to the vessel wall; asterisk indicates the clot; red staining demonstrates the presence of AA RBCs within the clot. *P < .05, **P < .01, ***P < .001. Ex, exchange; gluta, glutaraldehyde; WB, whole blood.
Next, we investigated if reduced deformability of sickle RBCs prevents their extrusion from the clot during retraction. Glutaraldehyde is commonly used to reduce deformability of RBCs.37,38 Low concentrations of glutaraldehyde decreased flexibility of normal RBCs at low shear stress, comparable to that observed in sickle RBCs (Figure 5B). Importantly, exposure of normal RBCs to glutaraldehyde before recombining with platelet-rich plasma significantly reduced the number of RBCs extruded into the serum during ex vivo clot contraction (Figure 5C).
RBC exchange transfusion is an effective therapy for certain acute and chronic complications of SCD.39,40 Because our data from the mouse model of SCD suggested that sickle RBCs contribute to the differences observed during clot contraction, we investigated if therapeutic RBC exchange in SCD patients affects the extrusion of RBCs during clot contraction. Before RBC exchange, the average percentage of HbA in the blood of sickle cell patients oscillated around 50%, as a consequence of previous exchange (supplemental Table 1). RBC exchange resulted in a further increase in the ratio of HbA to HbS, without affecting the total number of RBCs (supplemental Table 1). The number of RBCs released from preexchange clots was significantly increased compared with the number released from clots of sickle patients who were not on an RBC exchange protocol; however, the number was still lower than that extruded from nonsickle clots (Figure 5D). Furthermore, no additional increase in the number of released RBCs was observed between pre- and postexchange clots (Figure 5D). Importantly, analysis of Hb content in the pre- and postexchange clots demonstrated the presence of HbA, indicating incorporation of transfused normal RBCs within these clots (Figure 5E-F). Together, these data indicate that RBC exchange in SS patients leads to a long-lasting, partial normalization of RBC extrusion during clot contraction.
AA RBCs incorporate into venous clots formed in sickle mice
To model the effect of RBC exchange on ex vivo thrombus formation in sickle cell patients, we injected AA RBCs (supplemental Figure 4A) labeled with DiD lipophilic tracer into SS mice who were then subjected to the electrolytic injury thrombosis model. Strong DiD signal was detected within the clots, indicating the incorporation of AA RBCs into clots formed in the femoral veins of sickle mice (Figure 5G). Leakage of DiD dye into other cells was excluded by an in vitro experiment demonstrating a negligible number of double-stained RBCs after co-incubation of 2 separate populations of RBCs stained with either DiD or incubated with RBC specific antibody (supplemental Figure 4B).
Antisickling compounds do not reverse the entrapment of sickle RBCs in clots
Hydroxyurea (HU) reduces painful crises and hypercoagulability in sickle cell patients.41-44 To determine if HU treatment affects sickle RBC entrapment during clot retraction, we compared the number of RBCs extruded from ex vivo whole blood clots obtained from sickle cell patients receiving HU treatment or not. No significant differences were observed between these 2 groups of patients, either at baseline or following addition of the FXIIIa inhibitor (Figure 6A). Next, we investigated effects of the heterocyclic aldehyde, 5-hydroxmethyl-2-furfural (5HMF), an experimental agent that increases Hb oxygen affinity, thereby reducing RBC sickling and hypoxia-induced mortality in sickle mouse models of SCD.45 Treatment of SS mice with 5HMF at the dose used in the previously mentioned study did not significantly affect the number of RBCs extruded from clots formed ex vivo (Figure 6B). A combination of in vivo 5HMF treatment of mice and ex vivo inhibition of their blood with FXIIIa resulted in a nonsignificant increase in the number of extruded RBCs (Figure 6B). In addition, in vivo treatment of SS mice with N-acetyl cysteine, using doses previously shown to improve vascular responses in these mice,46 did not affect extrusion of RBCs from clots (supplemental Figure 5).
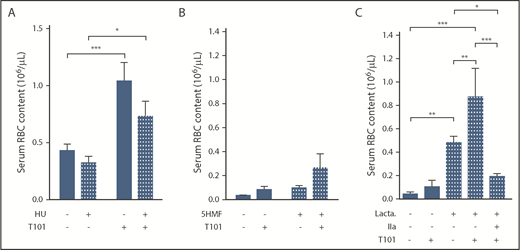
Effect of antisickling agents and lactadherin treatment on RBC extrusion from ex vivo whole blood clots. Number of RBCs extruded from clots, in the absence (−) or presence (+) of FXIIIa inhibitor T101 (20 µM), obtained from sickle cell patients receiving (n = 12) or not (n = 11) HU treatment (A) with SS mice treated in vivo with 5HMF (100 mg/kg of body weight; oral gavage; n = 13) or saline (n = 6) (B) or whole blood from SS mice treated ex vivo with phosphatidylserine inhibitor, lactadherin (1 µM; n = 3-10) (C). *P < .05, **P < .01, ***P < .001. IIa, thrombin.
Effect of antisickling agents and lactadherin treatment on RBC extrusion from ex vivo whole blood clots. Number of RBCs extruded from clots, in the absence (−) or presence (+) of FXIIIa inhibitor T101 (20 µM), obtained from sickle cell patients receiving (n = 12) or not (n = 11) HU treatment (A) with SS mice treated in vivo with 5HMF (100 mg/kg of body weight; oral gavage; n = 13) or saline (n = 6) (B) or whole blood from SS mice treated ex vivo with phosphatidylserine inhibitor, lactadherin (1 µM; n = 3-10) (C). *P < .05, **P < .01, ***P < .001. IIa, thrombin.
Phosphatidylserine contributes to the retention of RBCs in sickle clots via a thrombin-dependent mechanism
Increased expression of phosphatidylserine on the surface of circulating RBCs is well documented in sickle cell patients. Consistent with this observation, increased phosphatidylserine expression was also observed in mouse sickle RBCs, predominantly within the immature, CD71 positive population (supplemental Figure 6A-B). In a pilot experiment, the phosphatidylserine inhibitor lactadherin prevented tissue factor–initiated clot formation in AA but not SS blood (supplemental Figure 7). Furthermore, ex vivo addition of lactadherin changed the gross morphology of mouse SS clots and increased the number of extruded RBCs (supplemental Figure 7; Figure 6C). Interestingly, although T101 treatment alone did not affect the number of sickle RBCs extruded from clots, the combination of T101 and lactadherin resulted in an additive and statistically significant RBC release compared with lactadherin alone (Figure 6C). Stimulation of blood with thrombin reversed the effects of combined FXIIIa and phosphatidylserine inhibition (Figure 6C).
The cellular fraction of blood increases resistance of human sickle clots to fibrinolysis
Fibrinolysis is an important and integral part of the hemostatic system.47 To evaluate fibrinolysis in patients with SCD, we determined the fibrinolytic effect of tissue plasminogen activator (tPA) on clots formed in PPP or in whole blood using a ball sedimentation assay (supplemental video 1). We found that the clot lysis time was not different between control and SCD PPP samples at any tPA concentration (Figure 7A). Interestingly, however, compared with control whole blood clots, clot lysis time was significantly prolonged in clots formed in sickle whole blood. This difference was particularly apparent at low tPA concentrations (0.625 and 1.25 nM; Figure 7B). Furthermore, sickle trait whole blood clots demonstrated an intermediate phenotype in response to tPA (Figure 7B). Collectively, these data suggest the cellular fraction of blood contributes to resistance of sickle clots to fibrinolysis.
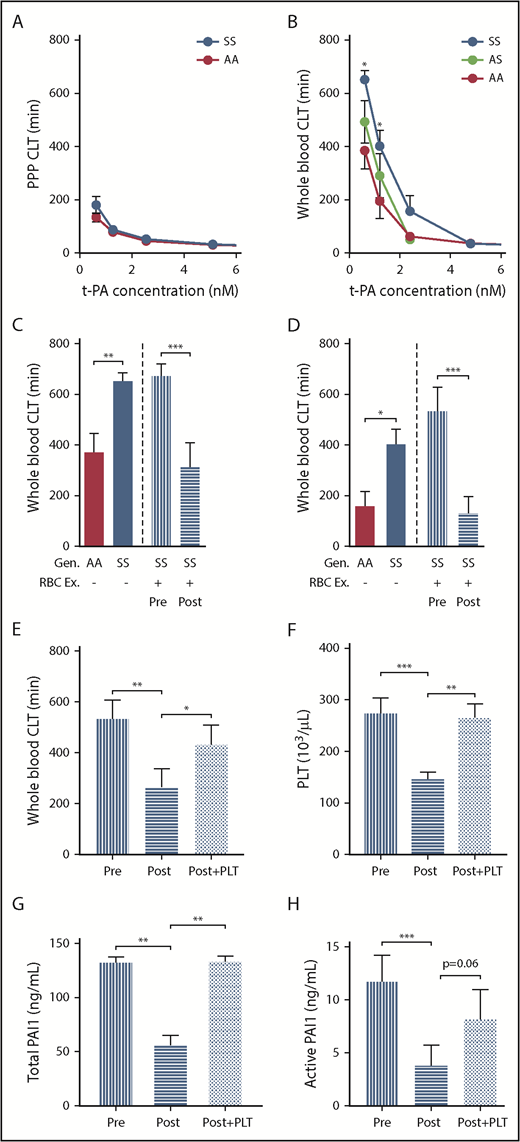
Resistance of sickle whole blood clots to tPA-mediated fibrinolysis is transiently prevented by RBC exchange in sickle patients. CLT of clots formed from PPP (A) or whole blood (B) of AA subjects (n = 8-9), AS (n = 11), and SCD (n = 13-15). Effect of tPA at 0.625 nM (C) and 1.25 nM (D) concentration on the CLT of clots formed from the whole blood of AA control subjects, SS patients, or SS patients undergoing RBC Ex pre- and postprocedure (n = 15). The values for the AA and SS groups. (C-D) The first 2 bars are copied from panel B. Effect of restoring PLT number in postexchange sample on CLT (E), whole blood PLT number (F), and serum levels of total (G) and active PAI-1 (H). All 4 parameters were analyzed in an independent cohort of sickle cell patients undergoing RBC exchange (n = 9-14), which was different from that used for the data presented in panels C and D. *P < .05, ** P< .01, ***P < .001.
Resistance of sickle whole blood clots to tPA-mediated fibrinolysis is transiently prevented by RBC exchange in sickle patients. CLT of clots formed from PPP (A) or whole blood (B) of AA subjects (n = 8-9), AS (n = 11), and SCD (n = 13-15). Effect of tPA at 0.625 nM (C) and 1.25 nM (D) concentration on the CLT of clots formed from the whole blood of AA control subjects, SS patients, or SS patients undergoing RBC Ex pre- and postprocedure (n = 15). The values for the AA and SS groups. (C-D) The first 2 bars are copied from panel B. Effect of restoring PLT number in postexchange sample on CLT (E), whole blood PLT number (F), and serum levels of total (G) and active PAI-1 (H). All 4 parameters were analyzed in an independent cohort of sickle cell patients undergoing RBC exchange (n = 9-14), which was different from that used for the data presented in panels C and D. *P < .05, ** P< .01, ***P < .001.
Red blood cell exchange in sickle patients transiently normalizes differences observed in tPA-mediated fibrinolysis between sickle and nonsickle clots
To determine if sickle RBCs mediate the resistance of whole blood clots to tPA, we investigated the effect of red cell exchange on tPA-mediated fibrinolysis. Clot lysis times in the preexchange whole blood clots were similar to that in sickle patients who did not undergo red cell exchange (Figure 7C-D). However, red cell exchange significantly reduced clot lysis times of postexchange whole blood clots (Figure 7C-D). We next investigated the mechanism responsible for the transient enhancement of fibrinolysis observed shortly after RBC exchange. We noticed that RBC exchange not only increased the percentage of HbA, as expected, but also significantly reduced platelet number in postexchange compared with preexchange blood samples (supplemental Table 2). We hypothesized that the reduced platelet number enhanced fibrinolysis in postexchange clots by reducing PAI-1 levels. Consistent with the initial observation demonstrated in Figure 7D, shortening of the clot lysis time (CLT) in postexchange compared with preexchange clots (Figure 7E) corresponded with reduced platelet numbers in the blood (Figure 7F) as well as reduced total (Figure 7G) and active (Figure 7H) PAI-1 serum levels between these 2 groups. Importantly, restoring platelet number in postexchange samples to that observed in preexchange blood (Figure 7F) resulted in significant prolongation of CLT compared with CLT observed in postexchange clots (Figure 7E), an effect that was associated with similar increases of total and active PAI-1 levels (Figure 7G-H).
Discussion
A chronic hypercoagulable state and increased risk of thrombotic complications in SCD are well-documented.2,11 The novel data presented in this study indicate that SCD may elicit these effects by dramatically changing the properties of the clot itself. The most striking observation was the increased rate and extent of fibrin deposition within venous clots in SS mice. Enhanced thrombin generation increases fibrin network density,48 providing 1 possible explanation for these observations. However, it is unclear whether the increased thrombin activity in sickle blood stems from increased expression of leukocyte tissue factor,3,49 plasma or platelet hypercoagulability,50 and/or increased RBC-mediated thrombin generation.51-53 Further studies are warranted to interrogate these possibilities. Interestingly, it has been demonstrated that sickle RBCs induce more fibrin clustering around themselves within fibrin clots formed ex vivo,31 which may in part explain the very dense fibrinlike structure surrounding RBCs that we observed in venous clots from SS mice.
Venous clots are often referred to as red thrombi because of the presence of large numbers of RBCs.18 We have demonstrated that sickle RBCs undergo extensive sickling within clots formed in the veins of SS mice. Importantly, the degree of RBC sickling was minimal in the blood pools surrounding the clots, suggesting that the sickling of RBCs was induced by hypoxic conditions within the venous thrombi and not by the experimental procedures during sample processing. Furthermore, these data imply that RBC sickling is triggered after incorporation into developing clots. Polymerization of HbS, which is followed by erythrocyte sickling, is promoted by hypoxia and acidosis in a time-dependent manner.54 Thus, this observation is particularly intriguing, because it implies that the extent of RBC sickling detected in anticoagulated whole blood vastly underestimates the degree of sickling that takes place in venous thrombi in SCD.
We next investigated if the morphologic change associated with RBC sickling affects clot contraction, which is an important phase of clot maturation.21,22 Several pieces of evidence support the possibility that changes in sickle RBC morphology mediate entrapment of these cells within the clot. First, electron microscopy demonstrated the presence of long, spiculelike processes extending from sickle RBCs that intertwined with each other and with the fibrin network (Figure 3D; supplemental Figure 3A). These processes, first described by White et al,55 may render sickled RBCs more resistant to extrusion during clot contraction. Second, glutaraldehyde-stiffened normal RBCs also showed increased retention within contracted clots (Figure 5C), suggesting that reduced RBC deformability decreases RBC extrusion during clot contraction. Finally, therapeutic RBC exchange in sickle cell patients produced a long-lasting effect on RBC extrusion from whole blood clots, shifting the response toward that observed in clots from healthy donors (Figure 6D). Importantly, the presence of HbA was observed not only in the whole blood but also in ex vivo clots generated from blood of sickle patients undergoing RBC exchange, indicating incorporation of donor RBCs (Figure 6E-F). This conclusion is also supported by the appearance of AA RBCs in venous clots formed in SS mice infused with normal mouse RBCs before induction of thrombosis (Figure 6G). RBC transfusion has been shown to reduce the incidence of stroke in sickle cell patients. Thus, it would be interesting to investigate if this therapeutic intervention also attenuates the previously proposed fragility of sickle clots by reducing the percentage of sickle RBCs in clots.
Interestingly, we observed that entrapment of sickle RBCs within clots was not significantly affected by 5HMF treatment of SS mice. The antisickling properties of 5HMF are mediated by increasing oxygen affinity to Hb. We speculate that, although 5HMF is sufficient to attenuate sickling of sickle RBCs in vitro and within the microcirculation,45 prolonged exposure of sickle RBCs to hypoxia and acidosis within the clot reduces effectiveness of the antisickling agent. Surprisingly, a similar number of RBCs was extruded during contraction of whole blood clots obtained from sickle cell patients irrespective of hydroxyurea treatment. This observation suggests that, although hydroxyurea can reduce the hypercoagulable state in sickle cell patients, as demonstrated by reduced markers of thrombin generation and fibrinolysis,41-44 it may not be as effective in modifying the properties of sickle clots once they are formed. However, these results should be interpreted with caution until the effect of hydroxyurea treatment on whole blood clot contraction can be assessed in a prospective clinical study or in mouse models of SCD, in which samples could be analyzed before and after hydroxyurea administration.
Of note, we observed that the entrapment of mouse sickle RBCs within whole blood clots was more pronounced than that observed with human samples. Furthermore, the entrapment of mouse sickle RBCs within clots was not affected by inhibition of FXIIIa activity, whereas FXIIIa inhibition partially enhanced extrusion of sickle RBCs from human clots. These species differences were associated with the appearance of “less sickling” on average of human compared with mouse sickle RBCs observed by electron microscopy (data not shown), suggesting that reduction in the intensity of sickling may favor FXIII-dependent release of RBCs from the clot. However, hydroxyurea treatment in a small cohort of SCD patients appeared to have no effect on extrusion of RBCs, either under basal conditions or after inhibition of FXIIIa, suggesting differences in the entrapment of mouse and human sickle RBCs are simply species-dependent.
In contrast to the lack of effect of antisickling agents, inhibition of phosphatidylserine by lactadherin significantly increased the extrusion of sickle RBCs and also unmasked the FXIIIa-dependent retention of RBCs in sickle mouse clots. The effect mediated by inhibition of phosphatidylserine was associated with an altered clot structure and was reversed by the addition of thrombin. These data indicate that retention of sickle RBCs during clot contraction is not only mediated by their abnormal morphology, but also by biochemical properties that promote thrombin generation, as we have previously demonstrated.51,56
To further investigate functional characteristics of sickle clots, we evaluated the resistance of preformed clots to fibrinolysis in control subjects and in sickle patient samples. Our findings demonstrate that the cellular components in sickle blood increase the resistance to tPA-mediated fibrinolysis because no difference between controls and sickle patients was observed in PPP. Similarly, we previously reported that the presence of RBCs and other cellular components in the blood of sickle patients was associated with increased thrombin generation that was not observed in platelet-free plasma.56 Furthermore, we and others have shown that increased thrombin generation leads to qualitative differences in the fibrin clot.48,57 Thus, the increased resistance of whole blood clots to tPA could be mediated, in part, by the presence of a denser fibrin network in sickle clots, as we demonstrated in sickle mice. In addition, elevated whole blood thrombin generation could lead to increased activation of thrombin-activatable fibrinolysis inhibitor,58 which could further inhibit fibrinolysis. Therapeutic RBC exchange resulted in reversal of susceptibility to fibrinolysis only in post- but not in preexchange clots, which was associated not only with further dilution of sickle Hb during the procedure, but also a 47% with a reduction in platelet count (supplemental Table 2) and serum PAI-1. Increased PAI-1 and prolongation of CLT observed after restoring platelet number in postexchange blood indicate RBC exchange normalizes the response to tPA-mediated fibrinolysis, at least in part, by reducing levels of platelet-derived PAI-1.
Epidemiologic studies have demonstrated that SCT is also associated with an increased risk of VTE59-61 and baseline evidence of activation of coagulation,62,63 albeit much less pronounced than that observed in SCD. This thrombotic propensity is presumably traceable to the heterozygous β-globin mutation, suggesting that SCT may represent an intermediate phenotype, at least with respect to VTE. In contrast to SCD, RBC sickling is not observed in the peripheral blood of SCT; however, it has been demonstrated that SCT RBCs may undergo sickling when exposed to extreme and prolonged hypoxia.64 Consistent with these observations, we observed an intermediate phenotype for AS mice with respect to venous clot fibrin content and resistance to tPA-mediated fibrinolysis of the clot formed from whole blood of sickle trait subjects. Further larger scale studies are required to determine whether biophysical conditions within developing venous thrombi are sufficient to promote Hb polymerization and sickling in sickle trait erythrocytes, and if so, whether such an event might lead to a heightened propensity to VTE in SCT.
Our data demonstrate that sickle RBCs alter the structure and dynamics of venous clot formation. Consistent with our results, Strauss et al reported that SCD alters the formation of polyhedrocytes within the core of the clot, which may affect clot stability.32 It is tempting to speculate that the structural changes observed in venous thrombi can render them more prone to embolization, which may explain, in part, the propensity for SCD and SCT patients with VTE to present with pulmonary embolism rather than deep vein thrombosis.8,11,59-61 Alternatively, the increased resistance of whole blood clots to fibrinolysis may enhance the likelihood of a symptomatic presentation with pulmonary embolism and/or pulmonary artery thrombi formed in situ. It is currently unknown whether long-term RBC exchange, which normalizes clot contraction and reverses resistance of sickle clots to fibrinolysis, mitigates the risk of VTE in SCD. Future clinical studies are needed to examine these possibilities.
For original data, please contact rafal_pawlinski@med.unc.edu.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors would like to thank Emeric Stauffer, Romain Fort, Giovanna Cannas, Philippe Connes, Alexandra Gauthier, Arnaud Hot and Solène Poutrel for their technical help with experiments and recruitment of patients and control subjects.
This study was supported by the National Institutes of Health, National Heart, Lung, and Blood Institute grants HL117659 (R.P., N.S.K., and K.I.A.), R01HL126974 (A.S.W.), T32 HL007149-35 (P.E.), and R01HL142604 (R.P.). The University of North Carolina Flow Cytometry Core Facility is supported in part by National Institutes of Health, National Cancer Institute (grant P30 CA016086) Cancer Center Core Support Grant to the University of North Carolina Lineberger Comprehensive Cancer Center
Authorship
Contribution: C.F. performed research, analyzed data, and wrote the paper; A.I., A.S., E.M.S., J.D.B., L. Buczek, M.W.H., L. Bhoopat, D.F.N., M.P., and B.C. performed research; W.B., P.E., Y.P., C.R., and K.I.A. contributed reagents and samples; N.S.K. and A.S.W., analyzed data and edited the paper; and R.P. designed research, analyzed data, and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Rafal Pawlinski, Division of Hematology/Oncology, Department of Medicine, 120 Mason Farm Rd, 1043 Genetic Medicine Building, Chapel Hill, NC 27599; e-mail: rafal_pawlinski@med.unc.edu.
