

Key Points
Constitutive NF-κB activation and blocked terminal differentiation trigger p53 signaling and antitumor immune escape mechanisms in ABC-DLBCL.
Simultaneous PD-1 blockade improves long-term efficacy of anti-CD20 immunotherapy in a multilesion preclinical mouse model of ABC-DLBCL.

Abstract
Refractory or relapsed diffuse large B-cell lymphoma (DLBCL) often associates with the activated B-cell-like (ABC) subtype and genetic alterations that drive constitutive NF-κB activation and impair B-cell terminal differentiation. Here, we show that DNA damage response by p53 is a central mechanism suppressing the pathogenic cooperation of IKK2ca-enforced canonical NF-κB and impaired differentiation resulting from Blimp1 loss in ABC-DLBCL lymphomagenesis. We provide evidences that the interplay between these genetic alterations and the tumor microenvironment select for additional molecular addictions that promote lymphoma progression, including aberrant coexpression of FOXP1 and the B-cell mutagenic enzyme activation-induced deaminase, and immune evasion through major histocompatibility complex class II downregulation, PD-L1 upregulation, and T-cell exhaustion. Consistently, PD-1 blockade cooperated with anti-CD20-mediated B-cell cytotoxicity, promoting extended T-cell reactivation and antitumor specificity that improved long-term overall survival in mice. Our data support a pathogenic cooperation among NF-κB-driven prosurvival, genetic instability, and immune evasion mechanisms in DLBCL and provide preclinical proof of concept for including PD-1/PD-L1 blockade in combinatorial immunotherapy for ABC-DLBCL.
Introduction
Activated B-cell-like diffuse large B-cell lymphomas (ABC-DLBCLs) are aggressive mature B-cell non-Hodgkin’s lymphomas that resemble the plasmablast stage of B-cell development, characterizing patients at high risk for relapse or failure to respond to R-CHOP standard of care (immunochemotherapy with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone).1-3 Recently, genomic analyses have revealed new outcome-associated genetically defined DLBCL subgroups,4,5 evidencing the additional genetic complexity that underlies the transcriptionally defined classification of DLBCL into germinal center B-cell (GCB)- and ABC-like subtypes.6,7 Yet, many of the genetic hallmarks of ABC-DLBCL pathogenesis ultimately converge in 2 main oncogenic pathways2,3,8-10 : activation of canonical NF-κB and impaired plasma cell terminal differentiation, with the latter being frequently the consequence of inactivating mutations/deletions of BLIMP1/PRDM111-13 or alternative genetic/epigenetic repressor mechanisms.13,14 Disruption of plasma cell gene signature has also been linked to FOXP1, which is an essential regulator of B-cell development15-17 and plasma cell programs.18,19
Mutations in TP53 are present in approximately 20% of DLBCLs4,20-24 and associate with poor survival in patients with DLBCL.20,25-27 The majority of TP53 mutations in human DLBCL are accompanied by loss of p53 function,20 where the expression of a mutant p53 protein may sometimes exert a dominant-negative regulation over any remaining wild-type p53 or acquire new oncogenic functions.28-30 Even though bi-allelic TP53 mutations are frequent in a distinct genetic subgroup of DLBCLs that show no ABC/GCB enrichment,4,21 alternative copy number-dependent mechanisms that affect other p53 pathway components and ultimately result in perturbed p53 signaling can be detected in 66% of newly diagnosed DLBCLs.31 For example, the negative modulator of p53 transcriptional activity, BCL2L12 (at 19q13.42), is amplified in a subset of DLBCLs,4,31 mainly comprising ABC-DLBC cases with cosegregated alterations in PRDM1/NF-κB modifiers and the highest contribution of activation-induced deaminase (AID)–driven signatures.4 Furthermore, mutated TP53 is a predictor of refractoriness or early relapse in DLBCL.32,33 Therefore, it is reasonable to expect that a fully functional p53 pathway is critical in all DLBCL types, and identification of novel therapeutic vulnerabilities will benefit from deeper understanding of the pathogenic cooperation among perturbed p53 signaling, aberrantly active NF-κB, and blockade of terminal B-cell differentiation in ABC-DLBCL.
Furthermore, it is becoming increasingly evident that DLBCL comprises not only the malignant large B cells but also a complex tumor microenvironment (TME) that may play a role in DLBCL progression and response to therapy.34 Negative selection checkpoints are required for removing autoreactive or aberrant GCBs,35,36 and it has been proposed that acquired somatic mutations harbored by malignant cells may remodel the TME and support survival.34 Here, we have explored the cross talk of genetic and TME deregulated mechanisms in the pathogenesis of DLBCL, unraveling NF-κB-driven molecular addictions and immunosuppressive signatures associated with responsiveness to immunotherapy in ABC-DLBCL.
Methods
Genetically modified mice
Mouse strains were obtained from the Jackson Laboratory, including p53F, Blimp1F, IKK2caGFPstopF, Cγ1-cre, and eYFPstopF. See supplemental Methods, available on the Blood Web site, for detailed information about strains, housing, immunizations, in vivo immunotherapy, and echography imaging. All animal care and procedures were approved by the Ethical Committee of Animal Experimentation of the University of Navarra and the Instituto de Salud Pública y Laboral de Navarra Health Department.
Human samples, primary cells, and cell lines
Normal fresh human tonsils and formalin-fixed paraffin-embedded samples from patients with DLBCL were studied with the approval of the Clinical Research Ethics Committee of the Clinica Universidad de Navarra and in accordance with ethical guidelines at the University Hospital of Katholieke Universiteit Leuven. See supplemental Methods for additional information regarding fresh cellular sorting, culture conditions of lymphoma cell lines, and the National Center for Biotechnology Information Gene Expression Omnibus data sets reanalyzed here.
Immunohistochemistry
Pathological analyses were performed using standard procedures and our previous experience,37 as detailed in supplemental Methods.
Transcriptomics and ChIP-seq analyses
Information regarding quantitative real-time polymerase chain reaction (qRT-PCR), RNA-seq, RNA interference, microarray expression, murine variable diversity joining (VDJ)-immunoglobulin heavy chain (IgH)-seq,38 and chromatin immunoprecipitation (ChIP)-seq, is detailed in supplemental Methods.
Flow cytometry and t-SNE analysis
Flow-based studies of surface and intracellular markers, gating strategies, Rphenograph clustering method, and t-distributed stochastic neighbor embedding (t-SNE) analysis were performed as detailed in supplemental Methods.
Statistical analyses
Statistical analyses were performed using GraphPad Prism v7.0 and are described in supplemental Methods.
Results
p53 surveillance protects GCBs from ABC-DLBCL lymphomagenesis induced by NF-κB activation and Blimp1 loss
Mice bearing compound mutations driving blockade of plasma cell terminal differentiation (by conditional deletion of Blimp1) and activation of canonical NF-κB (by conditional expression of IKK2ca, a constitutively active mutant form of IKK2) had been shown to develop lymphomas that resemble human ABC-DLBCL.9 Recurrently, a third oncogenic pathway is thought to promote additional genomic instability through impaired p53 signaling.21-23,31 To investigate the pathogenic cross talk of these 3 pathways, we crossed mice bearing conditional alterations in homologs of key human ABC-DLBCL mutations p53F/F, Blimp1F/F, IKK2caGFPstopF/stopF, and used the Cγ1-Cre to target conditional mutagenesis at early stages of the germinal center (GC) reaction (designated p, B, I, and C, respectively; Figure 1A).
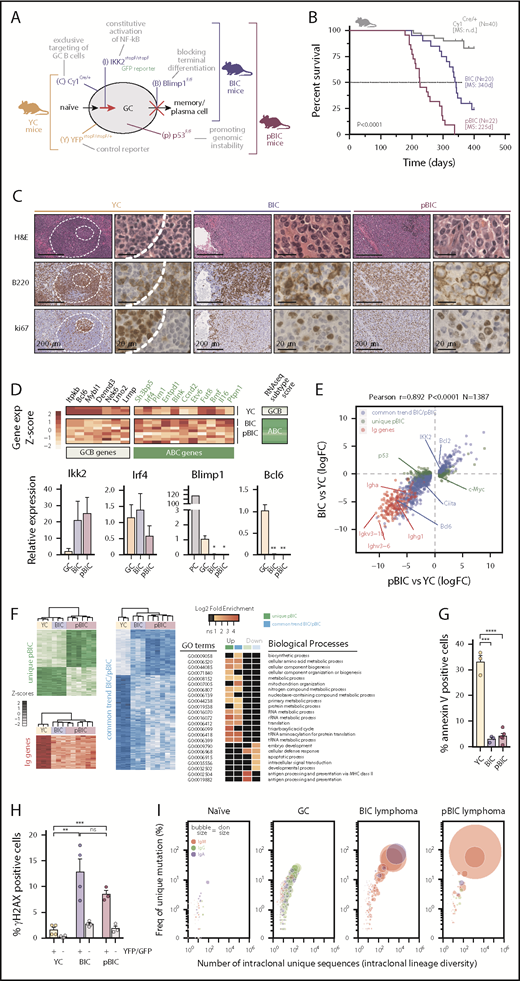
Conditional deletion of p53 cooperates with constitutive canonical NF-κB and Blimp1 loss in ABC-DLBCL lymphomagenesis. (A) Schematic diagram of the mutant mice and targeted B-cell functions used in this study. YC controls, YFPstopF/+Cγ1Cre/+; BIC, Blimp1F/FIKK2castopF/stopFCγ1Cre/+; pBIC, p53F/FBIC. (B) Overall survival of control or multilesion mice. (C) Representative immunohistochemical staining of hematoxylin and eosin, B220 and ki67 to label proliferating B cells in normal splenic GCs and murine diffuse B-cell lymphomas. Scale bars, 200 or 20 μm, as indicated. (D) RNA-seq gene expression classifier distinguishes ABC-DLBCL subtype in the murine lymphomas, which is confirmed by qRT-PCR of FACS-sorted reporter-positive normal GCBs or PCs, and lymphoma B cells (n≥3 animals). Relative values are normalized to GCB expression levels. (E) Scatter plot of differentially expressed genes (N = 1387) as measured by RNA-seq from GFP+/YFP+ reporter splenic B cells, showing log2 fold-changes in lymphoma relative to normal GCBs (n ≥ 3 animals). Genes were stratified and colored according to whether they were found in both lymphoma models or differentially expressed in the more aggressive pBIC model. (F) Heat maps of gene expression levels (left) and gene ontology (GO) analysis (right) for the categories of differentially expressed genes stratified in panel E. (G) Comparative percentages of apoptotic cells within reporter-positive control or lymphoma cells. (H) Comparative percentages of reporter-positive B cells that are positive for γH2AX by intracellular FACS. Gray bars represent YFP/GFP-negative normal cells from the same tumors. (I) Bubble plot illustrating the enrichment of VDJ-IgH clonal groups within reporter-positive murine control or lymphoma cells that accumulate unique somatic mutations (y-axis) in intraclonally diverse V sequences (x-axis). Bubble sizes represent the abundance of clonal barcoded single-molecule, and therefore clon size, whereas colors indicate the dominant isotype.
Conditional deletion of p53 cooperates with constitutive canonical NF-κB and Blimp1 loss in ABC-DLBCL lymphomagenesis. (A) Schematic diagram of the mutant mice and targeted B-cell functions used in this study. YC controls, YFPstopF/+Cγ1Cre/+; BIC, Blimp1F/FIKK2castopF/stopFCγ1Cre/+; pBIC, p53F/FBIC. (B) Overall survival of control or multilesion mice. (C) Representative immunohistochemical staining of hematoxylin and eosin, B220 and ki67 to label proliferating B cells in normal splenic GCs and murine diffuse B-cell lymphomas. Scale bars, 200 or 20 μm, as indicated. (D) RNA-seq gene expression classifier distinguishes ABC-DLBCL subtype in the murine lymphomas, which is confirmed by qRT-PCR of FACS-sorted reporter-positive normal GCBs or PCs, and lymphoma B cells (n≥3 animals). Relative values are normalized to GCB expression levels. (E) Scatter plot of differentially expressed genes (N = 1387) as measured by RNA-seq from GFP+/YFP+ reporter splenic B cells, showing log2 fold-changes in lymphoma relative to normal GCBs (n ≥ 3 animals). Genes were stratified and colored according to whether they were found in both lymphoma models or differentially expressed in the more aggressive pBIC model. (F) Heat maps of gene expression levels (left) and gene ontology (GO) analysis (right) for the categories of differentially expressed genes stratified in panel E. (G) Comparative percentages of apoptotic cells within reporter-positive control or lymphoma cells. (H) Comparative percentages of reporter-positive B cells that are positive for γH2AX by intracellular FACS. Gray bars represent YFP/GFP-negative normal cells from the same tumors. (I) Bubble plot illustrating the enrichment of VDJ-IgH clonal groups within reporter-positive murine control or lymphoma cells that accumulate unique somatic mutations (y-axis) in intraclonally diverse V sequences (x-axis). Bubble sizes represent the abundance of clonal barcoded single-molecule, and therefore clon size, whereas colors indicate the dominant isotype.
To investigate whether the IKK2-mediated constitutive activation of canonical NF-κB affected p53 in GC-derived B-cell lymphomas, we first examined p53 levels in the previously described mouse model that develop ABC-DLBCL9 (referred to as Blimp1F/FIKK2caGFPstopF/stopFCγ1Cre/+ [BIC] mice). These BIC lymphomas showed stabilization of the p53 protein (supplemental Figure 1A), as well as increased transcriptional p53 levels compared with normal GC cells (supplemental Figure 1B), suggesting ongoing DNA damage responses in the tumors. To further investigate the role of p53 in ABC-DLBCL progression, we conditionally deleted Trp53 in the multilesion BIC background (referred as pBIC mice; Figure 1A). These novel p53-deficient pBIC mice developed an ABC-DLBCL-like phenotype that was significantly more aggressive than the p53-proficient BIC mice (Figure 1B). In particular, pBIC mice exhibited more dramatic splenomegaly (supplemental Figure 1C) and malignant expansion of GFP+ lymphoma B cells (supplemental Figure 1D), corresponding to large cells (supplemental Figure 1E) that could not terminally differentiate into plasma cells (supplemental Figure 1F), and exhibited a B220+CD38+CD138−IgM+ immunophenotype (supplemental Figure 1G-I). Histologic examinations of splenic tumors showed morphological features resembling human DLBCL, characterized by disrupted architecture and a diffuse growth pattern of B220+ki67+ cells (Figure 1C). Consistently, a previously validated human RNA-seq-based subtype classifier39 distinguished the ABC-DLBCL phenotype in these murine tumors (Figure 1D, top), whereas qRT-PCR further confirmed the high expression of Ikk2 and Irf4 levels, as well as the loss of Blimp1 and Bcl6 expression (Figure 1D, bottom). Furthermore, hemizygous expression of IKK2ca resulted in delayed tumor onset and death in pBIC mice (supplemental Figure 2), indicating that p53-deficient lymphoma cells rely, at least in part, on the levels of active NF-κB signaling for tumor progression. Altogether, these results evidence a pathogenic cooperation between p53 loss and NF-κB activity, and suggest that p53 surveillance constitutes a main barrier to NF-κB-driven transformation of GCBs that fail to terminally differentiate.
Aberrant coexpression of FOXP1 and AID characterizes murine and human ABC-DLBCL
Next, we analyzed the transcriptional profile of BIC and pBIC tumors by RNA-seq (Figure 1E; supplemental Table 1). Both p53-proficient and p53-deficient murine ABC-DLBCLs demonstrated strong transcriptional similarity and loss of BCR diversity, indicating clonal enrichment. Upregulated genes revealed marked enrichment of metabolic and translation processes (Figure 1F), likely facilitating tumor growth. In contrast, downregulated genes appeared mainly enriched in processes related to development, intracellular signaling, or apoptosis (Figure 1F), which was consistent with the decreased cell death observed in these tumors (Figure 1G). We observed that murine ABC-DLBCL lymphomas accumulated high levels of unrepaired double-strand DNA breaks marked by γ-H2AX compared with control GCBs or normal lymphocytes from the same tumors (Figure 1H), and the archetypical IgH:cMyc translocation that is found in 5% to 15% of patients with DLBCL40,41 could be observed in the group of p53-deficient mice (supplemental Table 2). Although different mechanisms may be at the origin of chromosomal translocations in lymphoid malignancies,42 it is known that AID deregulation may lead to reciprocal chromosome translocations, including IgH:cMyc, facilitating the transformation of p53-deficient cells.43,44 Deep sequencing of murine lymphoma VDJ-IgH repertoires confirmed the strong clonal enrichment predicted by RNA-seq and demonstrated accumulation of additional somatic hypermutation (SHM) diversity compared with normal GCBs and unmutated resting B cells (Figure 1I; supplemental Table 3), suggesting the presence of ongoing AID activity in both p53-proficient and p53-deficient tumors. Moreover, we identified an aberrant coexpression of FOXP1 and the mutagenic enzyme AID in both BIC and pBIC lymphomas compared with normal GC or naive B cells from sheep red blood cells–immunized YFPstopF/+Cγ1Cre/+ (YC) control mice (Figure 2A-B; supplemental Table 4). Indeed, human tonsils and normal murine spleens confirmed the reciprocal expression of FOXP1 and AID during the GC transit of B cells (Figure 2C-D).
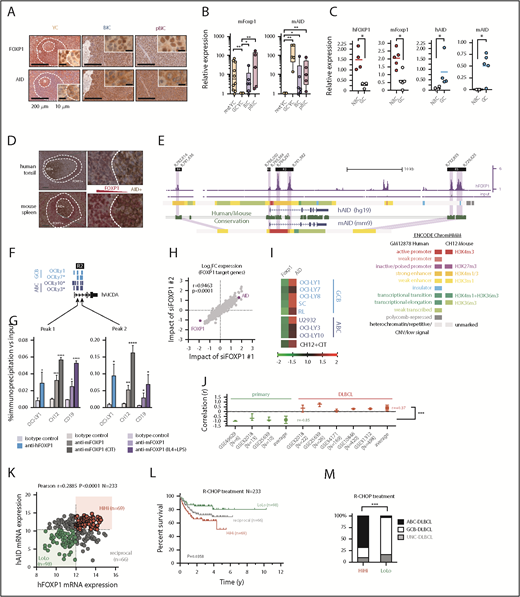
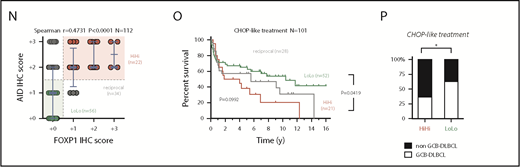
Aberrant coexpression of FOXP1 and AID characterizes NF-κB-driven murine and human ABC-DLBCL. (A) Representative immunohistochemical staining of FOXP1 (nuclear) and AID (cytoplasmic) in normal splenic GCs and lymphomas. Scale bars, 200 μm or 10 μm at insets. (B) Expression analysis by qRT-PCR of FACS-sorted reporter-positive normal resting or GCBs, and lymphoma B cells (n 5 animals). Relative values were normalized to resting or GCB expression levels for mFoxp1 or mAID, respectively. (C) Expression analysis by qRT-PCR of paired NBC or GCBs magnetically sorted from human tonsils or FACS sorted from normal murine spleens (n
5 animals). Relative values were normalized to resting or GCB expression levels for mFoxp1 or mAID, respectively. (C) Expression analysis by qRT-PCR of paired NBC or GCBs magnetically sorted from human tonsils or FACS sorted from normal murine spleens (n 4). Relative values are normalized to endogenous levels h/mGAPDH. (D) Representative images of inverse correlation of FOXP1 and AID expression in reactive human tonsil or murine spleen examined by IHC, using combined labeling of anti-FOXP1 (brown) and anti-AID (red). Scale bars, 200 μm; magnification, ×20. (E) ChIP-seq data from OCI-Ly1 cells showing enrichment of FOXP1 on the hAID locus. Distribution of FOXP1 occupancy is overlapped with chromatin-state models predicted by ENCODE ChromHMM from GM12878 (human B lymphoblastoid) or CH12 (mouse B-cell lymphoma) cells, and with the predicted conserved mAID gene locus according to Genome Browser comparison of hg19 and mm9 sequence data. Black boxes on top indicate genomic regions (R1-4) associated with transcriptional regulation of AID expression. (F) Comparison of hFOXP1 ChIP-seq peaks at intronic region 2 of the hAID locus observed here in OCI-Ly1 cells or previously in other GCB-DLBCL or ABC-DLBCL cell lines (GSE69009). (G) Validation of FOXP1 occupancy at AID intronic peaks and measured by ChIP-qPCR in human OCI-Ly1 DLBCL cells, as well as in resting or activated murine cells (ie, CH12 lymphoma cells and primary magnetically sorted CD19+ splenic cells). CIT, combination of anti-CD40 plus IL-4 and TNF-β; LPS, lipopolysaccharides. (H) Scatter plot of gene expression array data from OCI-Ly1 cells showing average log2 fold-changes (n = 3 replicates) in the expression of FOXP1-bound genes (according to ChIP-seq data from OCI-LY1) relative to scramble control after FOXP1 silencing with 2 different siRNAs. (I) Heat map of average fold-changes (n = 3 replicates) in the expression of FOXP1 and AID relative to scramble control after siRNA-mediated silencing of FOXP1 in DLBCL cell lines and in CIT-activated CH12 cells. (J) Forest graph plot of Pearson r coefficients measuring the correlation of hFOXP1 and hAID expression in previously published GSE series. (K) Scatter plot of FOXP1 and AID gene expression array data from R-CHOP-treated patients with DLBCL (n = 233, GSE10846). Median expression levels for FOXP1 (223287_s_at) and AID (219841_at) are indicated and were used as cutoff values for patient stratification. (L) Overall survival of R-CHOP-treated patients with DLBCL stratified by FOXP1/AID expression levels in panel K. (M) Distribution of COO-based subtypes in the FOXP1/AID HiHi and LoLo expression DLBCL subgroups stratified in panel K. COO subtypes were defined by expression signatures and were available in metadata from GSE10846. (N) Scatter plot of FOXP1 and AID protein expression data as measured by IHC scoring from CHOP-treated patients with DLBCL (n = 112). Median IHC scores are indicated and were used as cutoff values for patient stratification. (O) Overall survival of CHOP-treated patients with DLBCL stratified by FOXP1/AID IHC scores in panel N. (P) Distribution of COO-based subtypes in the FOXP1/AID HiHi and LoLo expression DLBCL subgroups stratified in panel N. COO subtypes were defined by the Hans IHC algorithm. HiHi, FOXP1highAIDhigh; LoLo, FOXP1lowAIDlow; NBC, naive B cells; UNC, unclassified.
4). Relative values are normalized to endogenous levels h/mGAPDH. (D) Representative images of inverse correlation of FOXP1 and AID expression in reactive human tonsil or murine spleen examined by IHC, using combined labeling of anti-FOXP1 (brown) and anti-AID (red). Scale bars, 200 μm; magnification, ×20. (E) ChIP-seq data from OCI-Ly1 cells showing enrichment of FOXP1 on the hAID locus. Distribution of FOXP1 occupancy is overlapped with chromatin-state models predicted by ENCODE ChromHMM from GM12878 (human B lymphoblastoid) or CH12 (mouse B-cell lymphoma) cells, and with the predicted conserved mAID gene locus according to Genome Browser comparison of hg19 and mm9 sequence data. Black boxes on top indicate genomic regions (R1-4) associated with transcriptional regulation of AID expression. (F) Comparison of hFOXP1 ChIP-seq peaks at intronic region 2 of the hAID locus observed here in OCI-Ly1 cells or previously in other GCB-DLBCL or ABC-DLBCL cell lines (GSE69009). (G) Validation of FOXP1 occupancy at AID intronic peaks and measured by ChIP-qPCR in human OCI-Ly1 DLBCL cells, as well as in resting or activated murine cells (ie, CH12 lymphoma cells and primary magnetically sorted CD19+ splenic cells). CIT, combination of anti-CD40 plus IL-4 and TNF-β; LPS, lipopolysaccharides. (H) Scatter plot of gene expression array data from OCI-Ly1 cells showing average log2 fold-changes (n = 3 replicates) in the expression of FOXP1-bound genes (according to ChIP-seq data from OCI-LY1) relative to scramble control after FOXP1 silencing with 2 different siRNAs. (I) Heat map of average fold-changes (n = 3 replicates) in the expression of FOXP1 and AID relative to scramble control after siRNA-mediated silencing of FOXP1 in DLBCL cell lines and in CIT-activated CH12 cells. (J) Forest graph plot of Pearson r coefficients measuring the correlation of hFOXP1 and hAID expression in previously published GSE series. (K) Scatter plot of FOXP1 and AID gene expression array data from R-CHOP-treated patients with DLBCL (n = 233, GSE10846). Median expression levels for FOXP1 (223287_s_at) and AID (219841_at) are indicated and were used as cutoff values for patient stratification. (L) Overall survival of R-CHOP-treated patients with DLBCL stratified by FOXP1/AID expression levels in panel K. (M) Distribution of COO-based subtypes in the FOXP1/AID HiHi and LoLo expression DLBCL subgroups stratified in panel K. COO subtypes were defined by expression signatures and were available in metadata from GSE10846. (N) Scatter plot of FOXP1 and AID protein expression data as measured by IHC scoring from CHOP-treated patients with DLBCL (n = 112). Median IHC scores are indicated and were used as cutoff values for patient stratification. (O) Overall survival of CHOP-treated patients with DLBCL stratified by FOXP1/AID IHC scores in panel N. (P) Distribution of COO-based subtypes in the FOXP1/AID HiHi and LoLo expression DLBCL subgroups stratified in panel N. COO subtypes were defined by the Hans IHC algorithm. HiHi, FOXP1highAIDhigh; LoLo, FOXP1lowAIDlow; NBC, naive B cells; UNC, unclassified.
Aberrant coexpression of FOXP1 and AID characterizes NF-κB-driven murine and human ABC-DLBCL. (A) Representative immunohistochemical staining of FOXP1 (nuclear) and AID (cytoplasmic) in normal splenic GCs and lymphomas. Scale bars, 200 μm or 10 μm at insets. (B) Expression analysis by qRT-PCR of FACS-sorted reporter-positive normal resting or GCBs, and lymphoma B cells (n5 animals). Relative values were normalized to resting or GCB expression levels for mFoxp1 or mAID, respectively. (C) Expression analysis by qRT-PCR of paired NBC or GCBs magnetically sorted from human tonsils or FACS sorted from normal murine spleens (n4). Relative values are normalized to endogenous levels h/mGAPDH. (D) Representative images of inverse correlation of FOXP1 and AID expression in reactive human tonsil or murine spleen examined by IHC, using combined labeling of anti-FOXP1 (brown) and anti-AID (red). Scale bars, 200 μm; magnification, ×20. (E) ChIP-seq data from OCI-Ly1 cells showing enrichment of FOXP1 on the hAID locus. Distribution of FOXP1 occupancy is overlapped with chromatin-state models predicted by ENCODE ChromHMM from GM12878 (human B lymphoblastoid) or CH12 (mouse B-cell lymphoma) cells, and with the predicted conserved mAID gene locus according to Genome Browser comparison of hg19 and mm9 sequence data. Black boxes on top indicate genomic regions (R1-4) associated with transcriptional regulation of AID expression. (F) Comparison of hFOXP1 ChIP-seq peaks at intronic region 2 of the hAID locus observed here in OCI-Ly1 cells or previously in other GCB-DLBCL or ABC-DLBCL cell lines (GSE69009). (G) Validation of FOXP1 occupancy at AID intronic peaks and measured by ChIP-qPCR in human OCI-Ly1 DLBCL cells, as well as in resting or activated murine cells (ie, CH12 lymphoma cells and primary magnetically sorted CD19+ splenic cells). CIT, combination of anti-CD40 plus IL-4 and TNF-β; LPS, lipopolysaccharides. (H) Scatter plot of gene expression array data from OCI-Ly1 cells showing average log2 fold-changes (n = 3 replicates) in the expression of FOXP1-bound genes (according to ChIP-seq data from OCI-LY1) relative to scramble control after FOXP1 silencing with 2 different siRNAs. (I) Heat map of average fold-changes (n = 3 replicates) in the expression of FOXP1 and AID relative to scramble control after siRNA-mediated silencing of FOXP1 in DLBCL cell lines and in CIT-activated CH12 cells. (J) Forest graph plot of Pearson r coefficients measuring the correlation of hFOXP1 and hAID expression in previously published GSE series. (K) Scatter plot of FOXP1 and AID gene expression array data from R-CHOP-treated patients with DLBCL (n = 233, GSE10846). Median expression levels for FOXP1 (223287_s_at) and AID (219841_at) are indicated and were used as cutoff values for patient stratification. (L) Overall survival of R-CHOP-treated patients with DLBCL stratified by FOXP1/AID expression levels in panel K. (M) Distribution of COO-based subtypes in the FOXP1/AID HiHi and LoLo expression DLBCL subgroups stratified in panel K. COO subtypes were defined by expression signatures and were available in metadata from GSE10846. (N) Scatter plot of FOXP1 and AID protein expression data as measured by IHC scoring from CHOP-treated patients with DLBCL (n = 112). Median IHC scores are indicated and were used as cutoff values for patient stratification. (O) Overall survival of CHOP-treated patients with DLBCL stratified by FOXP1/AID IHC scores in panel N. (P) Distribution of COO-based subtypes in the FOXP1/AID HiHi and LoLo expression DLBCL subgroups stratified in panel N. COO subtypes were defined by the Hans IHC algorithm. HiHi, FOXP1highAIDhigh; LoLo, FOXP1lowAIDlow; NBC, naive B cells; UNC, unclassified.
FOXP1 binding was detected by ChIP-seq (Figure 2E; supplemental Table 5) in all 4 previously known regulatory regions within the AID locus,45 which are well conserved between human and mice, and are predicted to insulate or modulate enhancer activity by ChromHMM analysis in available human and mouse ENCODE data (Figure 2E). Of note, both ABC- and GCB-like DLBCL human cell lines demonstrated direct binding of FOXP146 downstream of AID TSS at 2 discrete intronic peaks (Figure 2F). This FOXP1-to-AID locus binding was already evident in murine primary CD19+ resting B cells and the B-cell lymphoma CH12 cells that can be activated in vitro to induce AID expression, and significantly increased on cytokine stimulation (Figure 2G), suggesting a modulated commitment of FOXP1 in the insulation of the AID locus. Consistently, siRNA-mediated silencing of hFOXP1 followed by microarray mRNA profiling in DLBCL cells (Figure 2H), or by qRT-PCR in a cohort of mouse and human lymphoma cells lines (Figure 2I), demonstrated upregulation of AID in all scenarios. In contrast, multiple human DLBCL data sets revealed a consistent reversion of the negative correlation normally observed in primary samples (Figure 2J). A clinical relevance for FOXP1/AID aberrant coexpression was further supported when the concurrence of these 2 factors was analogously evidenced in retrospective analysis of human primary DLBCL samples (Figure 2K-N; supplemental Figure 3), and found that this was associated with reduced overall survival in R-CHOP and CHOP-treated patients with DLBCL (Figure 2L,O), predominantly of the ABC subtype (Figure 2M,P). These observations suggest that constitutive NF-κB and pro-oncogenic FOXP1 may cooperate with AID-triggered mutagenesis to promote ABC-DLBCL pathogenesis.
Mechanisms of immune evasion facilitate ABC-DLBCL tumor progression
B cells are themselves antigen-presenting cells that express major histocompatibility complex class II (MHC-II),47 and loss of MHC-II expression characterizes ABC-DLBCL.48 Consistently, RNAseq showed that MHC-II transactivator Ciita was significantly downregulated in both BIC and pBIC murine lymphomas (Figure 3A), which might impair T-cell activation. This was accompanied by a general trend toward loss of MHC-II gene expression (Figure 3B), which was most clear in p53-deficient lymphomas, in line with previous evidence for a link between p53 and MHC-I/II expression.49,50 Indeed, GO analysis revealed that antigen presentation via MHC-II was a biological process significantly downregulated in p53-deficient pBIC tumors (Figure 1F). To further investigate immunosurveillance in these NF-κB-driven lymphomas, we measured PD-L1 levels by flow cytometry and observed a clear increase of PD-L1 in the surface of the GFP+ lymphoma cells from both BIC and pBIC mice compared with GCB counterparts from YC control mice (Figure 3C). These results resembled human ABC-DLBCL primary lymphomas, which also showed increased PD-L1 expression and loss of MHC-II transactivator CIITA expression compared with GCB-DLBC cases (Figure 3A,C).
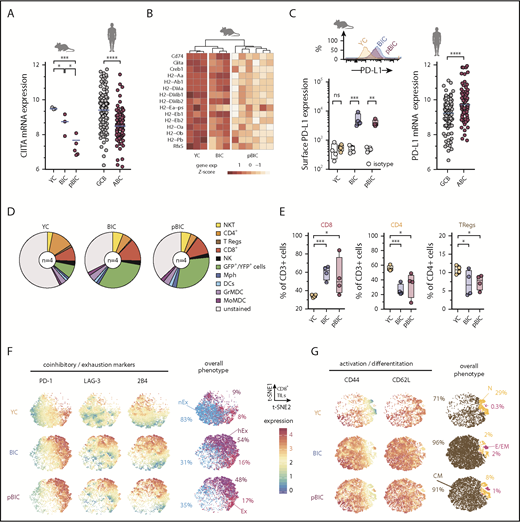
Decreased MHC-II gene expression and immune checkpoint deregulation cooperate with ABC-DLBCL genetic hallmarks to promote immune evasion. (A) Comparative gene expression levels of the MHC-II transactivator CIITA in GFP+/YFP+ reporter murine B cells from control or tumoral spleens (left) and human DLBCL (right, GSE10846). (B) Heat map of gene expression levels for MHC-II genes (KEGG pathway mmu04612) in GFP+/YFP+ reporter murine B cells from control or tumoral spleens. (C) Representative FACS histograms and comparative levels of surface PD-L1 in GFP+/YFP+ reporter murine B cells (left), and PD-L1 gene expression levels in human DLBCL (right, GSE10846). (D) Pie charts showing average percentages (n = 4 animals) of different immune cells in the control or tumoral spleens. (E) Comparative percentages of different T-cell populations in the control or tumoral spleens of indicated mice. (F-G) Multidimensional depiction by t-SNE of surface marker levels in aggregated total CD8+ T cells (dots) from control or tumoral spleens (n = 4 animals). Expression by FACS of coinhibitory and exhaustion markers (F) or activation and differentiation markers (G) were overlaid onto the overall t-SNE maps. Interpretation of the overall phenotype for each CD8+ T cell is color coded and annotated as nonexhausted (nEx; PD-1negLAG-3neg2B4neg), exhausted (Ex; PD-1hi), hyperexhausted cells that coexpress multiple inhibitory receptors besides PD-1 (hEx), naive (N; CD44lowCD62L+), central memory (CM; CD44highCD62L+), and effector or effector memory (E/EM; CD44highCD62Lneg). Percentages indicate average abundance of CD8+ T cells with the different exhaustion/differentiation phenotypes in YC control spleens or BIC/pBIC lymphomas.
Decreased MHC-II gene expression and immune checkpoint deregulation cooperate with ABC-DLBCL genetic hallmarks to promote immune evasion. (A) Comparative gene expression levels of the MHC-II transactivator CIITA in GFP+/YFP+ reporter murine B cells from control or tumoral spleens (left) and human DLBCL (right, GSE10846). (B) Heat map of gene expression levels for MHC-II genes (KEGG pathway mmu04612) in GFP+/YFP+ reporter murine B cells from control or tumoral spleens. (C) Representative FACS histograms and comparative levels of surface PD-L1 in GFP+/YFP+ reporter murine B cells (left), and PD-L1 gene expression levels in human DLBCL (right, GSE10846). (D) Pie charts showing average percentages (n = 4 animals) of different immune cells in the control or tumoral spleens. (E) Comparative percentages of different T-cell populations in the control or tumoral spleens of indicated mice. (F-G) Multidimensional depiction by t-SNE of surface marker levels in aggregated total CD8+ T cells (dots) from control or tumoral spleens (n = 4 animals). Expression by FACS of coinhibitory and exhaustion markers (F) or activation and differentiation markers (G) were overlaid onto the overall t-SNE maps. Interpretation of the overall phenotype for each CD8+ T cell is color coded and annotated as nonexhausted (nEx; PD-1negLAG-3neg2B4neg), exhausted (Ex; PD-1hi), hyperexhausted cells that coexpress multiple inhibitory receptors besides PD-1 (hEx), naive (N; CD44lowCD62L+), central memory (CM; CD44highCD62L+), and effector or effector memory (E/EM; CD44highCD62Lneg). Percentages indicate average abundance of CD8+ T cells with the different exhaustion/differentiation phenotypes in YC control spleens or BIC/pBIC lymphomas.
The imbalance of immune cell populations in the TME of murine ABC-DLBCLs may provide an additional explanation for tumor progression. Although the myeloid compartment did not exhibit evident relative changes compared with control YC spleens (Figure 3D; supplemental Figure 4A), the expansion of GFP+ lymphoma B cells was accompanied by a significant enrichment of CD8+ cells at the expense of CD4+ and CD25+FOXP3+ T regulatory cells (Figure 3E). Consistent with our hypothesis that infiltrating effector CD8+ cells became progressively exhausted through interaction with tumor PD-L1+ cells, most CD8+ T cells upregulated 1 or multiple inhibitory receptors including PD-1, LAG-3, and 2B4 (Figure 3F), which would decrease T-cell cytotoxicity against tumor cells.51 Therefore, this immune checkpoint phenotype in murine ABC-DLBCLs recapitulates the positive correlation between PD-L1 expression in the lymphoma cells and the presence of PD-1+ tumor-infiltrating lymphocytes (TILs) that has been observed in some human DLBCL studies.52-54 Furthermore, most tumor-associated CD8+ TILs coexpressed both CD44 and CD62L markers (Figure 3G), which are indicative of central memory phenotypes55 and suggest that these are tumor cognate cells with the potential to be reactivated. All these observations support the notion that typical ABC-DLBCL genetic alterations can ultimately cross talk with the TME to promote T-cell dysfunction through PD-L1/PD-1 signaling and weaken antigen presentation, therefore facilitating tumor immune evasion.
Immune checkpoint PD-1 blockade enhances anti-CD20 efficacy in the mouse ABC-DLBCL-like model
The presence of PD-1+ TILs is observed in 40% to 60% of human DLBCLs, even though its clinical value is still controversial.54 This prompted us to investigate whether our murine model system might support a predictive value of PD-L1+ tumor cells and PD-1+ TILs as biomarkers for successful combinational immunotherapy responses in ABC-DLBCL. A recent phase 2 study with anti-PD-1 monotherapy (Nivolumab)56 has shown low overall response rates in relapsed/refractory DLBCL, but there is limited information about the efficacy of PD-1 in combination with anti-CD20 immunotherapy in DLBCL. Therefore, we hypothesized that targeting DLBCL tumors with anti-CD20 monoclonal antibodies (mAbs) and simultaneous immune checkpoint blockade might be efficacious and tolerable. Combination immunotherapy with anti-CD20 and anti-PD-1 was superior to either monotherapy in overall survival responses, even though no significant improvement could be observed with the single anti-PD-1 regimen (Figure 4A). Consistently, ultrasound transversal measurements of the spleen during the first 3 cycles of treatment demonstrated that anti-CD20 as monotherapy or in combination was able to reduce the splenomegaly associated with the pBIC DLBCL, which was not evident with anti-PD-1 alone (Figure 4B).
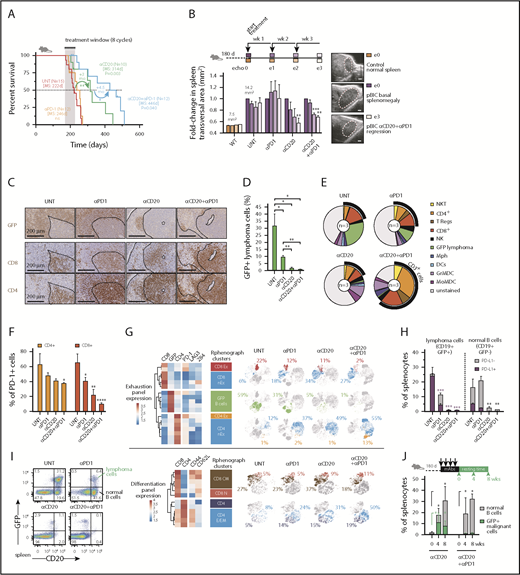
Immunotherapy with anti-PD-1 enhances anti-CD20 efficacy in the aggressive immunocompetent ABC-DLBCL mouse model. (A) Overall survival of murine ABC-DLBCL pBIC mice treated with different immunotherapy combinations. (B) Relative changes in splenic transversal areas measured by ventral ultrasound of pBIC mice (>180 days, with evidence of splenomegaly, n = 3) at 4 sequential times during immunotherapy treatment, as indicated in the scheme at the top. Representative ultrasound sections of the spleen are shown on the right. Scale bars, 1 mm. (C) Representative immunohistochemical staining of GFP, CD4, and CD8 to label T-cell infiltration in murine pBIC lymphomas (>180 days) that had received 4 weeks of immunotherapy. Scale bars, 200 μm. (D) Comparative percentages of splenic B220+GFP+ lymphoma cells from pBIC lymphomas (>180 days) in response to different 4-week immunotherapy regimens (n 3). (E) Pie charts showing percentages of immune cells in the spleen pBIC mice (>180 days) after 4-week immunotherapy (n = 3). (F) Comparative fractions of PD-1-positive T cells in the TME of pBIC mice (>180 days) after 4-week immunotherapy (n = 3). (G) Heat maps of mean expression intensity of coinhibitory/exhaustion (top) and activation/differentiation (bottom) surface markers within lymphocyte subsets detected by RPhenograph clustering (n = 3 mice × 4 groups). Distribution of these cell populations in response to 4-week immunotherapy (n = 3) is represented in t-SNE maps and colored. Percentages indicate average abundance of each cell population in the corresponding treatment group. (H) Comparative fractions of PD-L1-positive or PD-L1-negative cells within the compartment of lymphoma cells (CD19+GFP+) or neighbor normal B cells (CD19+GFP−) from pBIC mice (>180 days) after 4-week immunotherapy (n = 3). (I) FACS analysis to assess the specific depletion of lymphoma or normal B cells in the spleen of pBIC mice (>180 days) after 4-week immunotherapy (n = 3). (J) Percentages of reappearing splenic lymphoma cells (CD20+GFP+) or neighbor normal B cells (CD20+GFP−) during a resting period of 8 weeks after anti-CD20–based immunotherapy that can efficiently deplete the B-cell compartment in pBIC mice (>180 days, n
3). (E) Pie charts showing percentages of immune cells in the spleen pBIC mice (>180 days) after 4-week immunotherapy (n = 3). (F) Comparative fractions of PD-1-positive T cells in the TME of pBIC mice (>180 days) after 4-week immunotherapy (n = 3). (G) Heat maps of mean expression intensity of coinhibitory/exhaustion (top) and activation/differentiation (bottom) surface markers within lymphocyte subsets detected by RPhenograph clustering (n = 3 mice × 4 groups). Distribution of these cell populations in response to 4-week immunotherapy (n = 3) is represented in t-SNE maps and colored. Percentages indicate average abundance of each cell population in the corresponding treatment group. (H) Comparative fractions of PD-L1-positive or PD-L1-negative cells within the compartment of lymphoma cells (CD19+GFP+) or neighbor normal B cells (CD19+GFP−) from pBIC mice (>180 days) after 4-week immunotherapy (n = 3). (I) FACS analysis to assess the specific depletion of lymphoma or normal B cells in the spleen of pBIC mice (>180 days) after 4-week immunotherapy (n = 3). (J) Percentages of reappearing splenic lymphoma cells (CD20+GFP+) or neighbor normal B cells (CD20+GFP−) during a resting period of 8 weeks after anti-CD20–based immunotherapy that can efficiently deplete the B-cell compartment in pBIC mice (>180 days, n 3). MS, median survival; UNT, untreated.
3). MS, median survival; UNT, untreated.
Immunotherapy with anti-PD-1 enhances anti-CD20 efficacy in the aggressive immunocompetent ABC-DLBCL mouse model. (A) Overall survival of murine ABC-DLBCL pBIC mice treated with different immunotherapy combinations. (B) Relative changes in splenic transversal areas measured by ventral ultrasound of pBIC mice (>180 days, with evidence of splenomegaly, n = 3) at 4 sequential times during immunotherapy treatment, as indicated in the scheme at the top. Representative ultrasound sections of the spleen are shown on the right. Scale bars, 1 mm. (C) Representative immunohistochemical staining of GFP, CD4, and CD8 to label T-cell infiltration in murine pBIC lymphomas (>180 days) that had received 4 weeks of immunotherapy. Scale bars, 200 μm. (D) Comparative percentages of splenic B220+GFP+ lymphoma cells from pBIC lymphomas (>180 days) in response to different 4-week immunotherapy regimens (n3). (E) Pie charts showing percentages of immune cells in the spleen pBIC mice (>180 days) after 4-week immunotherapy (n = 3). (F) Comparative fractions of PD-1-positive T cells in the TME of pBIC mice (>180 days) after 4-week immunotherapy (n = 3). (G) Heat maps of mean expression intensity of coinhibitory/exhaustion (top) and activation/differentiation (bottom) surface markers within lymphocyte subsets detected by RPhenograph clustering (n = 3 mice × 4 groups). Distribution of these cell populations in response to 4-week immunotherapy (n = 3) is represented in t-SNE maps and colored. Percentages indicate average abundance of each cell population in the corresponding treatment group. (H) Comparative fractions of PD-L1-positive or PD-L1-negative cells within the compartment of lymphoma cells (CD19+GFP+) or neighbor normal B cells (CD19+GFP−) from pBIC mice (>180 days) after 4-week immunotherapy (n = 3). (I) FACS analysis to assess the specific depletion of lymphoma or normal B cells in the spleen of pBIC mice (>180 days) after 4-week immunotherapy (n = 3). (J) Percentages of reappearing splenic lymphoma cells (CD20+GFP+) or neighbor normal B cells (CD20+GFP−) during a resting period of 8 weeks after anti-CD20–based immunotherapy that can efficiently deplete the B-cell compartment in pBIC mice (>180 days, n3). MS, median survival; UNT, untreated.
To further investigate how the immunological landscape responded to these immunotherapy combinations, tumor spleens treated for 4 weeks were subjected to immune cell analysis. After anti-CD20 treatment, either as monotherapy or in combination with PD-1 blockade, we could observe a substantial depletion of GFP+ lymphoma B cells by immunohistochemistry (IHC; Figure 4C) or by fluorescence-activated cell sorting (FACS; Figure 4D). In addition, although anti-PD-1-alone or anti-CD20-alone treatment of lymphomas had limited effect, the combination of anti-CD20/anti-PD-1 displayed major changes in splenic cell populations (Figure 4E; supplemental Figure 4B). Of note, the overall population of CD3+ cells, including NKT, CD4+, T regulatory cells, and CD8+ cells, was markedly enriched in the anti-CD20/PD-1 combo immunological landscape, evidencing a distinctive T-cell-inflamed microenvironment that might promote extended therapeutic protection after treatment. Indeed, the combo treatment sustained a trend toward the clearance of PD-1+ T cells in the spleen (Figure 4F), in favor of CD8 and CD4 populations with absence or low levels of coinhibitory receptors PD-1, LAG3, or 2B4 (Figure 4G, top). Furthermore, these T cells preferentially expressed CD44 receptors, and although CD8 T cells conserved their prevalent initial central memory phenotype (CD44+CD62L+), new infiltrating CD4 T cells exhibited effector memory (CD44+CD62L−) phenotypes (Figure 4G, bottom), supporting the notion that an anti-CD20/anti-PD-1 combination was most efficient at establishing an overall activated T-cell microenvironment in the spleen. Consistent with this, maintained depletion of CD8 or CD4 T cells abrogated the survival benefit of combined immunotherapy in pBIC mice (supplemental Figure 5), indicating that T-cell-related immune-regulatory mechanisms mediate therapeutic effects in this ABC-DLBCL preclinical model.
Reinvigoration of the exhausted TILs was parallel to the specific destruction of PD-L1+CD19+GFP+ lymphoma B cells, whereas neighbor normal B cells remained largely negative for PD-L1 and were refractory to anti-PD-1 effects, succumbing only to the unspecific pan-B cytotoxicity of anti-CD20 mAbs (Figure 4H). These observations prompted us to consider that antitumor specificity contributed to the anti-CD20/anti-PD-1 combination benefit, as treatment with anti-PD-1 mAbs exclusively affected GFP+ lymphoma cells, whereas anti-CD20 treatment was nonselective in eliminating virtually all normal and tumoral B cells from the spleen (Figure 4I). In fact, incipient antitumoral responses with more reticular GFP+ dispersion and enlarged CD4/CD8 T-cell areas could be already observed on anti-PD-1 treatment, which was far more evident in the combination treatment (Figure 4C). Indeed, this specific antitumoral activity triggered by anti-PD-1 had some effect by itself on the overall depletion of GFP+ lymphoma B cells (Figure 4D), although it failed to significantly improve overall survival and tumor regression (Figure 4A-B), indicating that the more dramatic B-cell depletion mediated by anti-CD20 maximizes the effect of PD-1 blockade. Furthermore, we observed that anti-PD-1 administration conferred extended protection against the relapse of GFP+ malignant B cells on full depletion of CD20+ B cells, whereas it preserved the desirable normal B-cell reappearance (Figure 4J). All together, these results are consistent with the better long-term survival responses observed in the combination treatment, supporting our hypothesis that immune checkpoint blockade cooperates with the direct depletion of GFP+CD20+ lymphoma cells.
Discussion
The combination of constitutively active IKK2 with biallelic inactivation of Blimp1 and Trp53 in the GCBs of pBIC mice represents a genetic background that models the predicted effects of multiple combinatorial somatic mutations and copy-number alterations that are found in human DLBCL, and ultimately converge in 3 ABC-DLBCL hallmark pathways: constitutive activation of canonical NF-κB, blockade of terminal differentiation, and perturbation of p53 signaling. In full accord with the results of Calado et al,9 canonical NF-κB and Blimp1 loss (the BIC mice) cooperate to promote ABC-DLBCL. However, the long latency and the increased p53 expression in the oligoclonal tumors from these mice suggested that p53 surveillance is a main barrier for the pathogenesis of this disease. Indeed, we found that conditional deletion of p53 in mouse GCBs strongly synergized with IKK2 activation and Blimp1 loss to promote GC-derived lymphomagenesis. Thus, our work suggests that additional oncogenic events are required for NF-κB-driven transformation of GC-experienced plasmablasts that fail to terminally differentiate, which are initially resolved by a proficient p53 DNA damage response pathway.
In the search for major NF-κB downstream players that may promote genomic instability and contribute to ABC-DLBCL progression, we identified an aberrant coexpression of the transcription factor FOXP1 and the mutagenic enzyme AID. A relevance for this positive correlation was further supported when the concurrence of these 2 factors was similarly evidenced in retrospective collections of human primary DLBCL samples, showing association with reduced overall survival and the ABC subtype. Expression of FOXP1 might be promoting DLBCL survival by diverse mechanisms, including potentiation of the Wnt/b-catenin signaling57 or repression of pro-apoptotic genes,18,46,58 which is consistent with the decreased apoptosis that we observed in the murine ABC-DLBCLs. Upregulation of FOXP1 was expected, as it is broadly recognized as a prognostic indicator and biomarker for human ABC-DLBCL.59-61 In fact, NF-κB activation through IKK2 has been shown to cooperate with FOXP1 to promote lymphoma survival.46 However, the presence of AID and ongoing SHM in the tumors were unanticipated, as we had initially predicted that these would be suppressed in the presence of FOXP1 because we and others have shown that FOXP1 is a transcriptional repressor during the GC reaction that can bind AID and other GC-related genes.17,18,61 Indeed, we confirmed the reciprocal expression of FOXP1 and AID during the GC transit of B cells in human tonsils and normal murine spleens, and found a direct binding of FOXP1 downstream of AID TSS at 2 discrete intronic peaks, which exhibited strong enhancer chromatin marks and had been previously associated with ubiquitous silencers.45,62 However, the transcriptional repression of AID by FOXP1 results inefficient in the presence of constitutive NF-κB activation and IRF4 expression, which characterize ABC-DLBCL and are known to be direct strong activators of AID.45,62-66 Our results support that AID is a bona fide target of FOXP1 repression, even though FOXP1 insulation of AID becomes inefficient in lymphoma cells with constitutive NF-κB activity.
Tight regulation of AID in activated GCBs is necessary to maintain genomic integrity and avoid AID-driven lymphomagenesis.67-70 Indeed, evidence for AID expression and the accumulation of AID-related mutations has been observed in human DLBCL.24,71-73 Here, murine lymphomas accumulated high levels of unrepaired double-strand DNA breaks marked by γ-H2AX and the archetypical IgH:cMyc translocation could be evidenced in the group of p53-deficient mice. These genetic alterations might be attributable to AID off-target activity74-77 and are consistent with a role for p53 eliminating cells bearing AID-induced translocations.42-44 Furthermore, VDJ-IgH-seq from murine AID-positive ABC-DLBCL lymphomas, regardless of p53 deficiency, demonstrated strong clonal enrichment and accumulation of additional somatic hypermutation (SHM) diversity compared with normal GCBs and unmutated resting B cells, thus suggesting the presence of ongoing AID activity in the tumors during malignant progression. On the basis of all these observations, we postulate that constitutive expression of AID in human ABC-DLBCL cooperates with the prosurvival pressure imposed by NF-κB activation to increase genomic instability and accelerate lymphoma evolution, especially in malignant cells with perturbed p53 signaling.
Finally, we could confirm a cross talk of the malignant DLBCL cells with the TME to enable immune evasion. Both murine ABC-DLBCL-like models (BIC/pBIC) demonstrated a function of constitutive canonical NF-κB activation and Blimp1 loss in promoting T-cell dysfunction in DLBCL through upregulation of PD-L1 and loss of MHC-II gene expression. The most clear downregulation of MHC-II genes was associated with the perturbation of p53 signaling in pBIC tumors, which might contribute at least in part to their worse outcome. Interestingly, surface or soluble PD-L1 expression78-82 and reduced MHC-II antigen presentation48,83 have been linked to poor clinical outcomes and ABC-DLBCL. Furthermore, FOXP1 is a direct repressor of MHC-II genes in ABC-DLBCL,18,84 and NF-κB has been shown to be important for PD-L1 expression85-87 and stabilization,88 providing underlying mechanisms to support our observations that a constitutive NF-κB/FOXP1 pathway cooperates with perturbed p53 signaling to promote immune escape in ABC-DLBCL. Consistent with this, tumor-infiltrating CD8+ T cells exhibited central memory and exhausted phenotypes, with upregulation of 1 or multiple inhibitory receptors including PD-1, LAG-3, and 2B4, which would decrease T-cell cytotoxicity against tumoral cells.51,55 All these observations suggest that BIC/pBIC mouse models are able to recapitulate the complex cross talk between intracellular NF-κB activation and antitumoral immune evasion in ABC-DLBCL, becoming useful preclinical models to examine both the biology of the human disease and new therapeutic approaches.
The immune checkpoint phenotype observed here in murine ABC-DLBCLs recapitulates the positive correlation between lymphoma PD-L1 expression and the presence of PD-1+ TILs that has been observed in some human studies,52,53 supporting a predictive value of PD-L1 and exhausted CD8+ TILs as biomarkers for combinational immunotherapy response in ABC-DLBCL. Although the efficacy of immune checkpoint blockade varies greatly between different subtypes of lymphoma,54,89,90 and relapsed/refractory DLBCL has shown low response rates to anti-PD-1 monotherapy,56 it may be hoped that some patients with lymphoma will be more likely to respond to the right combination immunotherapy. Preliminary results from combination immunotherapy targeting both CD20 and PD-1 in follicular lymphoma are encouraging (NCT02446457).91 However, it is yet unclear whether this combination may have clinical benefits in DLBCL, for the future results from ongoing trials of anti-PD-1 mAbs in combination with anti-CD20-based chemo-immunotherapy (NCT02541565, NCT03259529, and NCT03366272) that are highly expected in the future. In this context, our study provides in vivo preclinical evidence that PD-1 blockade cooperates with anti-CD20-mediated depletion of lymphoma cells to reshape the immunosuppressive TME and facilitate long-term antitumor responses in NF-κB-driven p53-deficient ABC-DLBCL. Future mechanistic studies will make it possible to explore whether the in vivo benefit of this combination responds to interacting or completely independent mechanisms of drug action.92 In the p53-deficient/NF-κB-driven mouse model for ABC-DLBCL, we observed that although anti-CD20 mAbs provide a basal robust elimination of tumor cells, simultaneous immune checkpoint blockade with anti-PD-1 mAbs confers extended antitumor efficacy by promoting immune cell infiltration, narrowing co-inhibitory signals, and unraveling long-term cognate antitumor specificity in the DLBCL tumor microenvironment. These findings support that immune checkpoints hold promising therapeutic potential in ABC-DLBCL and provide preclinical proof of concept for the clinical evaluation of incorporating anti-PD-1 to the current anti-CD20-based modalities as combination immunotherapy for ABC-DLBCL.
Data discussed in this publication are accessible through Gene Expression Omnibus SuperSeries accession number GSE116290.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Matthew D. Scharff, Silvo Conticello, and Richard Chahwan for the kind gift of cell lines and reagents; Silvestre Vicent and Vasco Barreto for access to the p53F and eYFPstopF mouse lines, respectively; Angel Panizo for pathology advice; Genentech for in vivo functional grade anti-mouse CD20 mAbs; and Kathleen Van den Eynde, Julie Morscio, Ana Zafra, Xavier Morales, and Fernando López Aznal for technical assistance. The authors are also indebted to members of animal facility, morphology unit, CIMALab Diagnostics, and University of Navarra Biobank.
This work was supported by grants from Fondo Europeo de Desarrollo Regional (FEDER)/Ministerio de Ciencia, Innovación y Universidades-Agencia Estatal de Investigación (SAF2013-45787-R, SAF2017-82309-R) and Gobierno de Navarra (GN-106/2014) and contributions from Fundación Obra Social La Caixa to S.R., who was funded as research fellow of the European Marie Curie (FP7-PIIF-2012-328177) and Ramón y Cajal programs (RYC-2014-16399), as well as from Instituto de Salud Carlos III with FEDER cofunding (CIBERONC, PI16/00581 “Una manera de hacer Europa”), Worldwide Cancer Research (WCR15-1322), and Fundacion Arnal Planelles (J.A.M.-C). Additional funding was received from the Fondazione Umberto Veronesi (D.B.); Chemotherapy Foundation grant LLS SCOR-7012-16 and National Institutes of Health, National Cancer Institute grant R01-CA187492 (A.M.); and National Institutes of Health, National Institute of General Medical Sciences grant 1R01GM111741 and National Institutes of Health, National Institute of Allergy and Infectious Diseases grant 1R01AI132507 (T.M.).
Authorship
Contribution: J.A.M.-C. and S.R. conceived the study; M.P., J.A.M.-C., and S.R. designed experiments, analyzed data, and cowrote the manuscript; M.P., M.M.-V., E.F.R., M.-J.G.-B., and S.R. performed experiments; C.P. contributed to the design and interpretation of immunotherapy experiments; S.H.-S., D.A., N.C., and J.J.L. designed and performed the characterization of TME by cytometry and contributed to its interpretation; X.S. and O.B. performed pathological analyses; D.B. constructed and sequenced VDJ-IgH libraries; S.M. and T.M. performed computational immunology analyses; E.G. performed bioinformatics analyses; A.S., J.I.M.-F., K.L.B., J.C., and A.M.-B. performed experiments related to FOXP1 and AID interaction; and A.M. contributed to the design and support of experiments, as well as to discussion of results.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Sergio Roa, 55 Pio XII Ave, 31008 Pamplona, Spain; e-mail: sroa@unav.es; and Jose Angel Martinez-Climent, 55 Pio XII Ave, 31008 Pamplona, Spain; e-mail: jamcliment@unav.es.
