

Key Points
PGE2-G was elevated in sensory neurons of transgenic mice with SCD. This derivative of 2-AG produced hyperalgesia in control mice.
Blocking production of PGE2-G in mice with SCD inhibited hyperalgesia, suggesting a novel approach to treat pain in the disease.

Abstract
Pain is a characteristic feature of sickle cell disease (SCD), 1 of the most common inherited diseases. Patients may experience acute painful crises as well as chronic pain. In the Berkley transgenic murine model of SCD, HbSS-BERK mice express only human hemoglobin S. These mice share many features of SCD patients, including persistent inflammation and hyperalgesia. Cyclooxygenase-2 (COX-2) is elevated in skin, dorsal root ganglia (DRG), and spinal cord in HbSS-BERK mice. In addition to arachidonic acid, COX-2 oxidizes the endocannabinoid 2-arachidonoylglycerol (2-AG) to produce prostaglandin E2 (PGE2)–glycerol (PGE2-G); PGE2-G is known to produce hyperalgesia. We tested the hypotheses that PGE2-G is increased in DRGs of HbSS-BERK mice and sensitizes nociceptors (sensory neurons that respond to noxious stimuli), and that blocking its synthesis would decrease hyperalgesia in HbSS-BERK mice. Systemic administration of R-flurbiprofen preferentially reduced production of PGE2-G over that of PGE2 in DRGs, decreased mechanical and thermal hyperalgesia, and decreased sensitization of nociceptors in HbSS-BERK mice. The same dose of R-flurbiprofen had no behavioral effect in HbAA-BERK mice (the transgenic control), but local injection of PGE2-G into the hind paw of HbAA-BERK mice produced sensitization of nociceptors and hyperalgesia. Coadministration of a P2Y6 receptor antagonist blocked the effect of PGE2-G, indicating that this receptor is a mediator of pain in SCD. The ability of R-flurbiprofen to block the synthesis of PGE2-G and to normalize levels of 2-AG suggests that R-flurbiprofen may be beneficial to treat pain in SCD, thereby reducing the use of opioids to relieve pain.
Introduction
Sickle cell disease (SCD), 1 of the most common inherited diseases, results from a point mutation in hemoglobin that causes the polymerization of hemoglobin S, giving red blood cells their characteristic sickle shape.1,2 Pain is a characteristic feature of SCD and patients may experience acute painful crises as well as chronic pain.3 Hemolysis, inflammation, vaso-occlusion, vasculopathy, ischemia-reperfusion injury, organ damage, and neuropathy each contribute to chronic pain, a hallmark feature of SCD.4,5 In addition to ongoing pain and hyperalgesia, severe pain during a vaso-occlusive crisis often requires hospitalization.3 Because inflammation is a key component of SCD, nonsteroidal anti-inflammatory drugs (NSAIDs) are used to treat pain in SCD, but NSAIDs as a monotherapy are not effective.6 Opiates are typically used to treat pain in SCD3,7,8 but their long-term use is hampered by common side effects including tolerance, constipation, renal toxicity, focal hepatic necrosis, acute chest syndrome, respiratory depression, and addiction.9-11 Thus, there is a need to understand the mechanisms that contribute to pain in SCD so that new and effective treatments can be developed.
Transgenic murine models of SCD, such as the Berkley model,12 have been used to study pain mechanisms in SCD.13,14 HbSS-BERK mice are homozygous for knockout of both murine α and β globins and carry a single copy of the linked transgenes for human α and βS globins. These mice express human α and βS globin chains (thus, human hemoglobin S), but no murine α or β globins, and share many features of SCD patients, including persistent inflammation4,15 and hyperalgesia.13,14,16,17 Furthermore, HbSS-BERK mice exhibited sensitized nociceptors, characterized by spontaneous activity and enhanced responses to mechanical, heat, and cold stimuli18,19 and sensitization of nociceptive spinal dorsal horn neurons.20 This pattern is consistent with lowered pain thresholds and hyperalgesia in patients with SCD.21-24
Several biochemical changes in the skin, dorsal root ganglia (DRGs), and spinal cord of HbSS mice have been identified,18,25,26 including an increase in cyclooxygenase-2 (COX-2).16 COX-2 is well known for its oxygenation of arachidonic acid (AA) and synthesis of prostaglandins (PGs), particularly PGE2, which contribute to nociceptor sensitization by interacting with specific PG receptors.27 However, COX-2 also oxidizes the endocannabinoids 2-arachidonoylglycerol (2-AG)28 and anandamide,29 leading to the production of PG glycerol esters (PG-Gs) and PG ethanolamides, respectively. Of the 2 endocannabinoids, 2-AG is more abundant30,31 and this pattern holds true for skin and DRGs.32 Moreover, COX-2 oxidizes 2-AG and AA with comparable efficiency,33 but 2-AG does not interfere with AA for oxidation by COX-2. Importantly, R-enantiomers of profens, especially that of flurbiprofen, preferentially inhibit oxygenation of 2-AG by COX-2 compared with AA.34 One of the PG-Gs, PG E2 (PGE2)–glycerol (PGE2-G), is pronociceptive and produces mechanical and thermal hyperalgesia following intraplantar administration.35 Because COX-2 is elevated in SCD, an increase in COX-2–dependent oxidation, and the production of PGE2-G, is likely to occur.
We therefore investigated the contribution of PGE2-G to hyperalgesia and nociceptor sensitization in HbSS-BERK mice. We tested the hypothesis that PGE2-G is increased in DRGs of HbSS-BERK mice and sensitizes nociceptors, and that blocking its synthesis with R-flurbiprofen would decrease hyperalgesia in sickle mice. Given the recent report that PGE2-G activated P2Y6 receptors,36 we also tested whether a P2Y6 receptor antagonist37 would decrease hyperalgesia in HbSS-BERK mice.
Methods
Mice
Adult (5- to 9-months old), male HbSS-BERK, HbAA-BERK, and C3H/He mice were used in the studies. Control HbAA-BERK mice are littermates of HbSS-BERK and therefore have the same mixed genetic background as HbSS-BERK, but exclusively express normal human hemoglobin A and no murine globins. C3H/He mice (National Cancer Institute, Bethesda, MD) were used to confirm biochemical mechanisms and drug specificity. All experimental protocols were approved by the University of Minnesota Animal Care and Use Committee.
Drugs
2-AG, PGE2-G, PGE2, and their deuterated analogs 2-AG-d5, PGE2-G-d5, and PGE2-d4 were purchased from Cayman Chemical and stock solutions were prepared in ethanol (10 mg/mL). Stock solutions of R-flurbiprofen and S-flurbiprofen (23 mg/mL; Cayman Chemical), as well as EP receptor antagonists SC51089 (EP1, 21.8 μmol/mL), AH6809 (EP1-EP3, 33.5 μmol/mL), L798106 (EP3,18.6 μmol/mL), and L161982 (EP4, 15.3 μmol/mL), as well as P2Y6 nucleotide receptor antagonist MRS2578 (20 mg/mL) were prepared in dimethyl sulfoxide (DMSO). Receptor antagonists were purchased from Tocris. For use in experiments, drugs were diluted in to their final concentration in saline (behavior and electrophysiology) or medium (biochemistry).
COX-2 assays
Samples of DRGs (20 per mouse) were collected from HbSS- and HbAA-BERK mice for determination of COX-2 protein using western blots (25 µg of protein per sample) as described previously by our laboratory32,38 ; β-actin was used as a loading control. The density of immunoreactive bands was determined using ImageJ software. COX-2 activity was measured in triplicate in samples of lumber DRGs (bilateral L2-L6 per mouse) using a commercial assay kit (Cayman Chemical) according to the manufacturer’s directions. Data were normalized to total protein within each sample and are expressed as units of COX-2 activity (units per milligram of protein).
Measurement of 2-AG, PGE2-G, and PGE2
DRGs (L2-L6 bilaterally) from HbAA and HbSS mice treated with R-flurbiprofen or vehicle were collected, and lipids were extracted as described previously in our laboratory,39 in the presence of 2-AG-d5, PGE2-G-d5, and PGE2-d4 as internal standards. Samples were analyzed by liquid chromatography–nanospray ionization–tandem mass spectrometry on a Quantiva triple quadrupole (Thermo Fisher Scientific) interfaced to a Dionex Ultimate 3000 rapid-separation liquid chromatography high-performance liquid chromatography system (Thermo Fisher Scientific) using a home-packed Luna C18 column (5 μm, 120 Å, 200 mm × 75 μm internal diameter; Phenomenex) at room temperature with a flow rate of 0.3 μL/min. Mobile phase A was 10 mM ammonium formate with 0.1% formic acid in a 70:30 water-to-acetonitrile ratio, and mobile phase B was 10 mM ammonium formate with 0.1% formic acid in a 90:10 acetonitrile-to-dH2O ratio. The gradient started with 25% B with a flow rate of 1 μL/min. The quantification was based on the area ratio of analytical standard to tested analyte. Data were normalized to total lipids (grams). Analyses were performed by the Analytical Biochemistry Core facility of the University of Minnesota Cancer Center.
Behavioral assays
Mechanical hyperalgesia
Hyperalgesia was defined as a decrease in paw withdrawal threshold (also referred to as allodynia) determined for each hind paw using calibrated von Frey monofilaments (Stoelting) according to the method of Chaplan et al40 or as an increase in the frequency of paw withdrawal in response to a von Frey monofilament with a bending force of 3.9 millinewtons (mN) as described previously in our laboratory.32,39 The 3.9-mN monofilament was applied 10 times for 2 seconds each time to random locations on the plantar surface of each hind paw, and the number of withdrawal responses evoked by the monofilament was counted and expressed as a percentage of total stimuli (withdrawal frequency). Mean withdrawal latencies were obtained from 3 trials separated by at least 2 minutes. Baseline measurements of sensitivity to the 3.9-mN monofilament were taken over a 3-day period prior to each experiment to establish a consistent pattern of hyperalgesia for HbSS-BERK sickle mice and to confirm that HbAA-BERK control mice did not exhibit high levels of withdrawal responses.
Heat hyperalgesia
Paw withdrawal latency was recorded in response to a radiant heat stimulus as described previously in our laboratory.17 Mice were placed on a glass platform, covered with glass containers, and habituated for at least 30 minutes. Radiant heat was applied to the plantar surface of the hind paw and paw withdrawal latency (PWL) was determined. Mean PWL was calculated from 3 trials. The intensity of the heat source was adjusted so that mice withdrew their hind paws at ∼9 seconds during baseline testing. A cutoff time of 16 seconds was chosen to avoid tissue damage.
Cold avoidance test
The procedure of Zappia et al26 was followed. After habituation for 20 minutes, animals were tested for a preference between 2 plates held at 23°C or 30°C. The assay parameters required systemic administration of drugs. Data are reported as the amount of time the animal spent on the 23°C plate. The individual who conducted behavioral assays was blinded to the treatment and genotype of the mice.
Electrophysiology
Electrophysiological recordings were made from single nociceptive primary afferent fibers from the tibial nerve in anesthetized mice as described previously.41 Nerve fascicles were teased apart and extracellular recordings were obtained only from single fibers discriminated according to amplitude and shape of their action potential. The receptive field (RF) of each fiber was identified using a calibrated suprathreshold von Frey monofilament (149 mN). Nociceptors were identified by their responses evoked by different types of mechanical stimuli including light touch (brush with cotton-tipped applicator) and mild pinch. Fibers responding to pinch but not light touch were studied. Mechanical thresholds were determined using von Frey monofilaments. Conduction velocity was determined by electrically stimulating the RF and measuring the conduction latency and the distance between the RF and the recording electrode. Fibers were initially classified according to their conduction velocity (CV): Aβ = CV > 13.6 m/s, Aδ = 13.6 ≥ CV ≥ 1.3 m/s and C-fibers = CV < 1.3 m/s. The mean CV for all C-fibers included in this study was 0.61 ± 0.04 m/s and the median mechanical response threshold was 19.6 mN (12.7, 39.2; 25th, 75th percentile).
Responses evoked by controlled noxious mechanical, heat, and cold stimuli applied to the RF were obtained. Mechanically evoked responses were obtained using calibrated von Frey monofilaments. Threshold (millinewtons) was defined as the minimum force needed to evoke a response in at least 50% of the trials. Evoked responses to a 149-mN von Frey monofilament applied for 5 seconds were expressed as the mean of 3 trials, each separated by 60 seconds. A Peltier device (5 mm2 probe) was used to deliver computer-controlled thermal stimuli to the RF. Heat (36°C to 50°C for 5 seconds) and cold stimuli (28°C to 0°C for 10 seconds) were applied from a base temperature of 32°C with 1 to 2 minutes between stimuli. Heat stimuli were applied in ascending increments of 2°C and cold stimuli were presented in descending increments of 4°C.
cAMP assay
Primary cultures of dissociated DRGs were prepared from ganglia dissected from all levels of the spinal cord of adult C3H/He male mice as described previously by our laboratory.39,42 The final cell suspension was plated at a density of 30 000 cells on a laminin-coated 24-well plate and maintained in supplemented Ham F12/Delbecco’s modified Eagle medium in a humidified atmosphere of 5% CO2 at 37°C for ∼24 hours. At the start of the experiment, the culture medium was replaced with vehicle, PGE2-G (10 μM), or PGE2 (10 μM) with or without a pool of EP receptor antagonists. Cells were incubated with drugs for 10 minutes at 37°C. After aspiration of solution, cells were frozen for 2 hours and then thawed in 0.5-mL cold enzyme-linked immunosorbent assay buffer. Cyclic adenosine monophosphate (cAMP) levels were quantified using a cAMP enzyme-linked immunosorbent assay kit (Cayman Chemical) according to the manufacturer’s protocol for the nonacetylated method. Each sample was assayed in triplicate, and data are expressed as a picomoles per well.
Statistical analyses
Normally distributed data are presented as mean ± standard error of the mean (SEM). These data were analyzed using parametric tests and 2-way analysis of variance (ANOVA) with repeated measures where applicable. Post hoc analyses were performed using Bonferroni or Fisher’s least significant difference post hoc tests to determine differences between specific groups. Data that did not pass a normality test are presented as median (25th percentile, 75th percentile) and were analyzed using Kruskal-Wallis 1-way ANOVA on ranks followed by Dunn or Tukey tests to determine differences between groups.
Results
COX-2, PGE2, and PGE2-G were elevated in DRGs of HbSS-BERK mice
COX-2 is constitutively expressed in the spinal cord of mice but not in DRGs.43,44 COX-2 protein, detected by western blot, was dramatically increased in DRGs collected from HbSS-BERK mice compared with those of HbAA-BERK mice (Figure 1A-B). Functionally, the level of COX-2 activity was almost 10-fold higher in HbSS- compared with HbAA-BERK mice (Figure 1C).
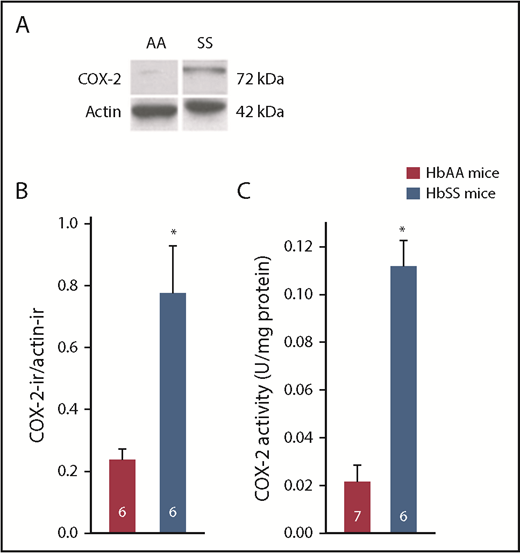
HbSS-BERK mice were characterized by increased expression of COX-2 protein and increased COX-2 activity in DRGs compared with HbAA-BERK mice. (A) Digitized images of a western blot of COX-2 and β-actin in DRGs (25 µg of protein per sample). Immunoblots were probed with rabbit anti–COX-2 (1/500; Cayman Chemical) and visualized with a peroxidase-conjugated goat anti-rabbit immunoglobulin G (IgG; Amersham Biosciences). Mouse anti-actin (1:20 000; Sigma-Aldrich) immunoreactivity within each sample, visualized with peroxidase-conjugated goat anti-mouse IgG, was used as a loading control. Images of the bands were cropped from different parts of the same gel to optimize visualization of immunoreactivity (ir) and space. (B) Summary of quantitative analysis digitized images of immunoblots. The amount of COX-2 protein is the ratio of COX-2–ir to actin-ir within the same sample. Data were transformed to log10 for parametric statistical analysis. (C) Activity of COX-2 was measured in 10 DRGs per mouse (L2-L6 bilaterally). Data were normalized to total protein within each sample and expressed as units of COX-2 activity per milligram of protein. Legend in panel C is common to panel B. Numbers in bars represent sample sizes. *Different from HbSS-BERK mice at P < .001; Student t test.
HbSS-BERK mice were characterized by increased expression of COX-2 protein and increased COX-2 activity in DRGs compared with HbAA-BERK mice. (A) Digitized images of a western blot of COX-2 and β-actin in DRGs (25 µg of protein per sample). Immunoblots were probed with rabbit anti–COX-2 (1/500; Cayman Chemical) and visualized with a peroxidase-conjugated goat anti-rabbit immunoglobulin G (IgG; Amersham Biosciences). Mouse anti-actin (1:20 000; Sigma-Aldrich) immunoreactivity within each sample, visualized with peroxidase-conjugated goat anti-mouse IgG, was used as a loading control. Images of the bands were cropped from different parts of the same gel to optimize visualization of immunoreactivity (ir) and space. (B) Summary of quantitative analysis digitized images of immunoblots. The amount of COX-2 protein is the ratio of COX-2–ir to actin-ir within the same sample. Data were transformed to log10 for parametric statistical analysis. (C) Activity of COX-2 was measured in 10 DRGs per mouse (L2-L6 bilaterally). Data were normalized to total protein within each sample and expressed as units of COX-2 activity per milligram of protein. Legend in panel C is common to panel B. Numbers in bars represent sample sizes. *Different from HbSS-BERK mice at P < .001; Student t test.
PGE2, 2-AG, and PGE2-G were quantified in DRGs to asses a functional significance for the increase in COX-2 (Table 1). Consistent with the increase in COX-2, levels of both PGE2 and PGE2-G were increased in HbSS- compared with HbAA-BERK mice, and the increase in PGE2-G was accompanied by a decrease in the level of 2-AG, a substrate for COX-2. Moreover, when HbSS-BERK mice were treated systemically with the COX-2 inhibitor R-flurbiprofen, production of PGE2-G was blocked and 2-AG increased whereas the level of PGE2 remained elevated. Consequences of blocking production of PGE2-G in HbSS-BERK mice on hyperalgesia were tested in vivo using behavioral and electrophysiological assays.
Levels of PGE2-G, 2-AG, and PGE2 in DRG of mice
| Lipid | HbAA-BERK, n = 8 | HbSS-BERK, n = 8 | HbSS + R-flurbiprofen, n = 6 |
|---|---|---|---|
| PGE2-G, fmol | 90 ± 12 | 592 ± 110* | 86 ± 17† |
| 2-AG, pmol | 569 ± 46 | 274 ± 35* | 873 ± 180† |
| PGE2, pmol | 1.8 ± 0.3 | 11.1 ± 1.3* | 9.5 ± 1.2‡ |
| Lipid | HbAA-BERK, n = 8 | HbSS-BERK, n = 8 | HbSS + R-flurbiprofen, n = 6 |
|---|---|---|---|
| PGE2-G, fmol | 90 ± 12 | 592 ± 110* | 86 ± 17† |
| 2-AG, pmol | 569 ± 46 | 274 ± 35* | 873 ± 180† |
| PGE2, pmol | 1.8 ± 0.3 | 11.1 ± 1.3* | 9.5 ± 1.2‡ |
DRGs were collected 60 minutes after intraperitoneal injection of vehicle (ratio of DMSO to Tween 80 to saline, 13.5%:0.5%:86%) or drug. HbAA-BERK mice were injected with vehicle; HbSS-BERK mice were injected with vehicle or R-flurbiprofen (9 mg/kg). Lipids of interest were normalized to the total lipid (grams) within each sample and are expressed as the mean ± SEM. Data were converted to log10 for parametric statistical analysis. n indicates the sample size.
Different from HbAA mice within the same lipid at P < .003.
Different from HbSS mice within the same lipid at P < .001 (1-way ANOVA, Bonferroni t test).
Different at P < .001.
R-flurbiprofen decreased hyperalgesia in HbSS-BERK mice
HbSS-BERK mice exhibited mechanical allodynia and cold hyperalgesia relative to HbAA-BERK mice as expected (Figure 2). Systemic administration of R-flurbiprofen reduced mechanical allodynia in a dose-dependent manner with a 50% effective dose of 6.5 mg/kg (Figure 2A). Moreover, the absence of an effect of the enantiomer S-flurbiprofen on mechanical allodynia in HbSS-BERK mice indicates that the effect is specific to the R-enantiomer of the drug (Figure 2B). Systemic administration of R-flurbiprofen (9 mg/kg, intraperitoneally) also reduced sensitivity to cold stimuli in HbSS-BERK at 60 minutes postinjection (Figure 2C). A peripheral site of action for R-flurbiprofen on mechanical hyperalgesia was confirmed following intraplantar administration of the drug (Figure 2D). The absence of an effect of R-flurbiprofen on mechanical and cold sensitivity following systemic and peripheral administration in HbAA-BERK mice (Figure 2B-D) indicates that the drug does not cause analgesia under the conditions used.
![Figure 2. R-flurbiprofen decreased mechanical hyperalgesia and cold sensitivity in HbSS-BERK mice. Mechanical hyperalgesia (allodynia) was defined as a decrease in mechanical withdrawal threshold of the hind paw. Littermates expressing normal human hemoglobin (HbAA-BERK) were used as the control. (A) R-flurbiprofen (intraperitoneal) decreased mechanical hyperalgesia in HbSS-BERK mice in a dose-dependent manner with an 50% effective dose of 6.54 ± 3.8 mg/kg (Prism software; GraphPad Software, Inc). For perspective, the open square represents the mean mechanical threshold in naive HbAA-BERK mice from panel D. *Different from vehicle (V; ratio of DMSO to Tween 80 to saline, 27%:0.5%:72.5%). Data were transformed to log10 for parametric statistical analysis. (F[3, 31] = 15.393; P < .001, 1-way ANOVA, N = 6-9 mice per group). (B) R-flurbiprofen (9 mg/kg intraperitoneally) acutely increased the mechanical threshold in HbSS-BERK, but not HbAA-BERK, mice. In addition, the same dose of S-flurbiprofen had no effect in HbSS-BERK mice. *Different from baseline (BL) and vehicle within HbSS-BERK mice (ratio of DMSO to Tween 80 to saline, 13.5%:0.5%:86%). #Different from S-flurbiprofen within HbSS-BERK mice at P < .001 (F[12, 99] = 4.71; P < .001, 2-way ANOVA repeated measures, treatment × time, N = 6-9 mice per group). (C) R-flurbiprofen (9 mg/kg intraperitoneally) reduced cold avoidance at 60 minutes post–drug injection. Data (median and 25% and 75% range) represent the amount of time mice spent on the colder plate (23°C). *Different from HbAA-BERK mice within vehicle treatment or naive condition. #Different from vehicle within HbSS-BERK mice P < .05, Kruskal-Wallis 1-way ANOVA (H[5] = 35.04 with Dunn multiple comparisons test; number within each bar represents sample size). (D) Intraplantar injection of R-flurbiprofen (30 μg/10 μL) suppressed mechanical allodynia in HbSS-BERK mice (F[9, 69] = 2.761; P = .008, 2-way ANOVA repeated measures, treatment × time, N = 5-8 mice per group) whereas the same dose of S-flurbiprofen or vehicle (ratio of DMSO to Tween 80 to saline, 13.5%:0.5%:86%), had no effect. *Different from HbAA-BERK mice within baseline (BL) at P < .001. #Different from other HbSS-BERK groups at 60 minutes and from BL within the same treatment group; P < .001; Bonferroni t test.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/133/18/10.1182_blood-2018-11-884346/3/m_blood884346f2.png?Expires=1765889840&Signature=qTdXMnw2s65PS7CwFGbBXi3WEA5fTethgl~FEg56YLx7AJdvGLb1RsaBhoOgOD9g5PQ3~1NKs2TI0y5a2spwatwbNbpwVefyU6IHoIvmmALYhY~EJ~qTVF2mmWkFhoYLYP8AWDAh8mNv0bjbyvJlzulYWlxe2HzkCwMb8TRGOGI65tvYWfwRU2M4RilozCUM99RooVDHcKpc-lbeEbAKqamBeEF9tyiJFjorp4~ghWEZN54yLnnH0lUNu3R7GQ4Ig4-rNBlWcdZAql6SmOb9s7U2vU3t9kt81Y0OofbccOgKCbARH4n8WQ4TaVdhp1d6wqKJAWjvYlukrtOq53xdvA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
R-flurbiprofen decreased mechanical hyperalgesia and cold sensitivity in HbSS-BERK mice. Mechanical hyperalgesia (allodynia) was defined as a decrease in mechanical withdrawal threshold of the hind paw. Littermates expressing normal human hemoglobin (HbAA-BERK) were used as the control. (A) R-flurbiprofen (intraperitoneal) decreased mechanical hyperalgesia in HbSS-BERK mice in a dose-dependent manner with an 50% effective dose of 6.54 ± 3.8 mg/kg (Prism software; GraphPad Software, Inc). For perspective, the open square represents the mean mechanical threshold in naive HbAA-BERK mice from panel D. *Different from vehicle (V; ratio of DMSO to Tween 80 to saline, 27%:0.5%:72.5%). Data were transformed to log10 for parametric statistical analysis. (F[3, 31] = 15.393; P < .001, 1-way ANOVA, N = 6-9 mice per group). (B) R-flurbiprofen (9 mg/kg intraperitoneally) acutely increased the mechanical threshold in HbSS-BERK, but not HbAA-BERK, mice. In addition, the same dose of S-flurbiprofen had no effect in HbSS-BERK mice. *Different from baseline (BL) and vehicle within HbSS-BERK mice (ratio of DMSO to Tween 80 to saline, 13.5%:0.5%:86%). #Different from S-flurbiprofen within HbSS-BERK mice at P < .001 (F[12, 99] = 4.71; P < .001, 2-way ANOVA repeated measures, treatment × time, N = 6-9 mice per group). (C) R-flurbiprofen (9 mg/kg intraperitoneally) reduced cold avoidance at 60 minutes post–drug injection. Data (median and 25% and 75% range) represent the amount of time mice spent on the colder plate (23°C). *Different from HbAA-BERK mice within vehicle treatment or naive condition. #Different from vehicle within HbSS-BERK mice P < .05, Kruskal-Wallis 1-way ANOVA (H[5] = 35.04 with Dunn multiple comparisons test; number within each bar represents sample size). (D) Intraplantar injection of R-flurbiprofen (30 μg/10 μL) suppressed mechanical allodynia in HbSS-BERK mice (F[9, 69] = 2.761; P = .008, 2-way ANOVA repeated measures, treatment × time, N = 5-8 mice per group) whereas the same dose of S-flurbiprofen or vehicle (ratio of DMSO to Tween 80 to saline, 13.5%:0.5%:86%), had no effect. *Different from HbAA-BERK mice within baseline (BL) at P < .001. #Different from other HbSS-BERK groups at 60 minutes and from BL within the same treatment group; P < .001; Bonferroni t test.
R-flurbiprofen decreased mechanical hyperalgesia and cold sensitivity in HbSS-BERK mice. Mechanical hyperalgesia (allodynia) was defined as a decrease in mechanical withdrawal threshold of the hind paw. Littermates expressing normal human hemoglobin (HbAA-BERK) were used as the control. (A) R-flurbiprofen (intraperitoneal) decreased mechanical hyperalgesia in HbSS-BERK mice in a dose-dependent manner with an 50% effective dose of 6.54 ± 3.8 mg/kg (Prism software; GraphPad Software, Inc). For perspective, the open square represents the mean mechanical threshold in naive HbAA-BERK mice from panel D. *Different from vehicle (V; ratio of DMSO to Tween 80 to saline, 27%:0.5%:72.5%). Data were transformed to log10 for parametric statistical analysis. (F[3, 31] = 15.393; P < .001, 1-way ANOVA, N = 6-9 mice per group). (B) R-flurbiprofen (9 mg/kg intraperitoneally) acutely increased the mechanical threshold in HbSS-BERK, but not HbAA-BERK, mice. In addition, the same dose of S-flurbiprofen had no effect in HbSS-BERK mice. *Different from baseline (BL) and vehicle within HbSS-BERK mice (ratio of DMSO to Tween 80 to saline, 13.5%:0.5%:86%). #Different from S-flurbiprofen within HbSS-BERK mice at P < .001 (F[12, 99] = 4.71; P < .001, 2-way ANOVA repeated measures, treatment × time, N = 6-9 mice per group). (C) R-flurbiprofen (9 mg/kg intraperitoneally) reduced cold avoidance at 60 minutes post–drug injection. Data (median and 25% and 75% range) represent the amount of time mice spent on the colder plate (23°C). *Different from HbAA-BERK mice within vehicle treatment or naive condition. #Different from vehicle within HbSS-BERK mice P < .05, Kruskal-Wallis 1-way ANOVA (H[5] = 35.04 with Dunn multiple comparisons test; number within each bar represents sample size). (D) Intraplantar injection of R-flurbiprofen (30 μg/10 μL) suppressed mechanical allodynia in HbSS-BERK mice (F[9, 69] = 2.761; P = .008, 2-way ANOVA repeated measures, treatment × time, N = 5-8 mice per group) whereas the same dose of S-flurbiprofen or vehicle (ratio of DMSO to Tween 80 to saline, 13.5%:0.5%:86%), had no effect. *Different from HbAA-BERK mice within baseline (BL) at P < .001. #Different from other HbSS-BERK groups at 60 minutes and from BL within the same treatment group; P < .001; Bonferroni t test.
Electrophysiological studies determined that R-flurbiprofen reduced sensitization of C-fiber nociceptors (N = 24) in HbSS-BERK mice (Figure 3). Intraplantar administration of R-flurbiprofen near the receptive field decreased spontaneous activity relative to vehicle from 60 to 120 minutes post–drug administration (Figure 3A,E). R-flurbiprofen increased the mechanical response threshold in HbSS-BERK mice as indicated by a 100% to 200% increase in threshold (Figure 3B). The increase in threshold was associated with a decrease in response to the suprathreshold von Frey stimulus of 149 mN (Figure 3C,F).
![Figure 3. R-flurbiprofen decreased sensitization of C-fiber nociceptors in HbSS-BERK mice. The 24 C-fibers included in this analysis had a mean CV of 0.61 ± 0.04 m/s. Intraplantar administration of R-flurbiprofen administration (30 mg/10 mL) (A) decreased the rate of spontaneous discharge in C-fiber nociceptors, (B) increased mechanical response thresholds, and (C) decreased responses to stimulation with a 149 mN von Frey monofilament. (A) F[4, 68] = 3.9, P < .001. *Different from vehicle at P < .05. #Different from time 0 (preinjection) at P < .05. (B) F[3, 57] = 2.8, P < .05. Different from vehicle at **P < .01, ***P < .005). (C) F[4, 52] = 13.1, P < .001. #Different from vehicle at **P < .005, ***P < .001. ###Different from time 0 (preinjection) at P < .001. Mechanical response thresholds (in millinewtons) were converted to percent change scores to enable parametric tests to be used for analysis of the drug effect. Data are mean ± SEM and were analyzed using 2-way ANOVA for repeated measures with the Bonferroni t test. Representative examples of responses of 2 C-fiber nociceptors before and after intraplantar administration of vehicle and R-flurbiprofen are shown in panels D-F. (D) Conduction latencies: multiple traces were overlapped to show consistency. The left arrow indicates the onset of the stimulus, and the right arrow indicates the action potential of the fiber of interest. (E) Spontaneous activity over a 45-second period for each condition. (F) Raw data demonstrating the response to a 149-mN stimulus over 5 seconds for each condition. Vertical bars indicate voltage scale in panels E-F: vehicle = 0.50 V; R-flurbiprofen = 0.25 V.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/133/18/10.1182_blood-2018-11-884346/3/m_blood884346f3.png?Expires=1765889840&Signature=iAjNubtwMBCvswSr3MlCrbW8SSTQcVFEoyCWGZ1tMGe5UkZn9yimux6V07ax9UYNy02r3LzRzSKyoru94NqTZ2gyRksBKr-q5wt3tcfjjuVPESLO~37zK-qX2XmRx1xEUyTuCl8GS41HnFF6D5jm4hMl2Ez9zCioaeYK43xqH8RNSHWbR2ONbidXrowt~uSjuMdXH~xKj06gScvPJpFAcYHKmCz59etlbb6gr6W1XJnyHn385UUNEIBSbEu8j8u3cyyiOBt9W2xh8wCPQGhFnq9sB4qGpttsHVwS0TAk5WTxlyJ5eLWPFWA-OS2XhP9aljAZUClQzizGzMddeK8E6w__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
R-flurbiprofen decreased sensitization of C-fiber nociceptors in HbSS-BERK mice. The 24 C-fibers included in this analysis had a mean CV of 0.61 ± 0.04 m/s. Intraplantar administration of R-flurbiprofen administration (30 mg/10 mL) (A) decreased the rate of spontaneous discharge in C-fiber nociceptors, (B) increased mechanical response thresholds, and (C) decreased responses to stimulation with a 149 mN von Frey monofilament. (A) F[4, 68] = 3.9, P < .001. *Different from vehicle at P < .05. #Different from time 0 (preinjection) at P < .05. (B) F[3, 57] = 2.8, P < .05. Different from vehicle at **P < .01, ***P < .005). (C) F[4, 52] = 13.1, P < .001. #Different from vehicle at **P < .005, ***P < .001. ###Different from time 0 (preinjection) at P < .001. Mechanical response thresholds (in millinewtons) were converted to percent change scores to enable parametric tests to be used for analysis of the drug effect. Data are mean ± SEM and were analyzed using 2-way ANOVA for repeated measures with the Bonferroni t test. Representative examples of responses of 2 C-fiber nociceptors before and after intraplantar administration of vehicle and R-flurbiprofen are shown in panels D-F. (D) Conduction latencies: multiple traces were overlapped to show consistency. The left arrow indicates the onset of the stimulus, and the right arrow indicates the action potential of the fiber of interest. (E) Spontaneous activity over a 45-second period for each condition. (F) Raw data demonstrating the response to a 149-mN stimulus over 5 seconds for each condition. Vertical bars indicate voltage scale in panels E-F: vehicle = 0.50 V; R-flurbiprofen = 0.25 V.
R-flurbiprofen decreased sensitization of C-fiber nociceptors in HbSS-BERK mice. The 24 C-fibers included in this analysis had a mean CV of 0.61 ± 0.04 m/s. Intraplantar administration of R-flurbiprofen administration (30 mg/10 mL) (A) decreased the rate of spontaneous discharge in C-fiber nociceptors, (B) increased mechanical response thresholds, and (C) decreased responses to stimulation with a 149 mN von Frey monofilament. (A) F[4, 68] = 3.9, P < .001. *Different from vehicle at P < .05. #Different from time 0 (preinjection) at P < .05. (B) F[3, 57] = 2.8, P < .05. Different from vehicle at **P < .01, ***P < .005). (C) F[4, 52] = 13.1, P < .001. #Different from vehicle at **P < .005, ***P < .001. ###Different from time 0 (preinjection) at P < .001. Mechanical response thresholds (in millinewtons) were converted to percent change scores to enable parametric tests to be used for analysis of the drug effect. Data are mean ± SEM and were analyzed using 2-way ANOVA for repeated measures with the Bonferroni t test. Representative examples of responses of 2 C-fiber nociceptors before and after intraplantar administration of vehicle and R-flurbiprofen are shown in panels D-F. (D) Conduction latencies: multiple traces were overlapped to show consistency. The left arrow indicates the onset of the stimulus, and the right arrow indicates the action potential of the fiber of interest. (E) Spontaneous activity over a 45-second period for each condition. (F) Raw data demonstrating the response to a 149-mN stimulus over 5 seconds for each condition. Vertical bars indicate voltage scale in panels E-F: vehicle = 0.50 V; R-flurbiprofen = 0.25 V.
PGE2-G generates hyperalgesia in HbAA-BERK control mice
The ability of PGE2-G to generate hyperalgesia was confirmed in control mice. Because sensitization of nociceptors occurs at the site of transduction of stimuli to the firing of action potentials, drugs were injected intraplantarly, near the site of behavioral and electrophysiological testing. PGE2-G (1 µg, intraplantarly) increased sensitivity (frequency of withdrawal) to the mechanical stimulus of 3.9 mN in the treated paw of HbAA-BERK mice from 30 to 90 minutes following injection (Figure 4A). A lower dose of PGE2-G (0.1 µg, intraplantarly) generated a twofold increase in frequency withdrawal at only the time point of 60 minutes (1-way ANOVA with repeated measures (F[5, 20] = 3.96, P = .012, N = 5 mice per group; data not shown). A pool of PG receptor antagonists (SC51089 [EP1], AH6809 [EP1-EP3], L798106 [EP3], and L161982 [EP4]) did not block the effect of PGE2-G, nor did it reduce mechanical hyperalgesia in HbSS-BERK mice (Figure 4B). The efficacy of this pool of antagonists in blocking PGE2 was confirmed in vivo and in vitro. Intraplantar injection of PGE2 in naive C3H/He mice increased mechanical sensitivity, and the effect was blocked by coadministration of the pool of PG receptor antagonists (Figure 4C). The pool of antagonists also blocked the generation of cAMP in response to PGE2 in primary cultures of dissociated DRGs from C3H/He mice (Figure 4D). It is noteworthy that PGE2-G did not generate cAMP at the concentration tested.
![Figure 4. PGE2-G produced hyperalgesia in HbAA-BERK mice. PGE2-G (2.3 nmol/10 µL, intraplantarly) was compared with the vehicle used for the pool of EP receptor antagonists (ratio of DMSO to ethanol to saline, 30%:1%:60%,10 µL, intraplantarly). (A) PGE2-G increased the frequency of withdrawal to a mechanical stimulus (3.9 mN). Coinjection of PGE2-G with vehicle or a pool of EP1, EP2, EP3, and EP4 receptor antagonists (SC51089, AH6809, L798106, and L161982, respectively, 20 nmol each, intraplantarly) did not reduce the effect of PGE2-G. Vehicle alone had no effect in HbAA-BERK mice. Two-way ANOVA for repeated measures demonstrated a significant treatment × time interaction (F[10, 106] = 2.67; P = .006, N = 8-9 mice per group). *Different from baseline (BL) and vehicle values at P < .004; Bonferroni t test. (B) The same pool of EP antagonists was ineffective in suppressing mechanical hyperalgesia in HbSS-BERK mice (F[1, 64] = 0.98; P = .326, 2-way ANOVA for repeated measures, N = 6-9 mice per group). (C) The efficacy of the pool of antagonists was validated against hyperalgesia evoked by PGE2 (9.8 nmol, intraplantarly) in naive C3H mice (2-way ANOVA for repeated measures (F[3, 80] = 8.88; P = .001, N = 5-6 mice per group). *Different from BL values at P < .013. #Different from PGE2 plus antagonists at P = .017; Bonferroni t test. (D) PGE2 increased cAMP in primary cultures of dissociated DRGs. The pool of EP receptor antagonists (10 μM per antagonist) blocked the effect of PGE2. Data are presented as picomoles of cAMP per well. *Different from other treatment groups and vehicle (V; ratio of DMSO 0.2% to ethanol 0.04% to saline 99.76%). Data were transformed to log10 for parametric statistical analysis. (F[3, 31] = 15.393; P < .001, 1-way ANOVA, N = 6 wells per treatment across 2 cultures).](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/133/18/10.1182_blood-2018-11-884346/3/m_blood884346f4.png?Expires=1765889841&Signature=3TgaFaJghWQ3zz4Q1ilL2gF7l5RtxOQYmFlQb0hbybIWq-HDyraQTDA66CWLBhh4VfvI0Ph2hB~7o-mBJs7f1-HFlKL6YItjUtbNMcYsKPdoTQR3Hoy9WhOX-vVKLx75o6xEg~srDNlI5e-1ym8aQ9S~9gC-qmBfWDaBciPPWFqCUu2llU65fyZFucnX8VgPs3veVmcBk4DZmkKXFpO79kYwjo31jJ8~mLziU-9EGUt1mqg29MJAa3kzV6zzP9KtezTGEqPl8KPYf~tn-5eWfy~j75uiP0SGUhDQdNMBsGVDP2twaLZ7dtpz4OQKfujLgesByFGmDHQDjJ9-MvlhoA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
PGE2-G produced hyperalgesia in HbAA-BERK mice. PGE2-G (2.3 nmol/10 µL, intraplantarly) was compared with the vehicle used for the pool of EP receptor antagonists (ratio of DMSO to ethanol to saline, 30%:1%:60%,10 µL, intraplantarly). (A) PGE2-G increased the frequency of withdrawal to a mechanical stimulus (3.9 mN). Coinjection of PGE2-G with vehicle or a pool of EP1, EP2, EP3, and EP4 receptor antagonists (SC51089, AH6809, L798106, and L161982, respectively, 20 nmol each, intraplantarly) did not reduce the effect of PGE2-G. Vehicle alone had no effect in HbAA-BERK mice. Two-way ANOVA for repeated measures demonstrated a significant treatment × time interaction (F[10, 106] = 2.67; P = .006, N = 8-9 mice per group). *Different from baseline (BL) and vehicle values at P < .004; Bonferroni t test. (B) The same pool of EP antagonists was ineffective in suppressing mechanical hyperalgesia in HbSS-BERK mice (F[1, 64] = 0.98; P = .326, 2-way ANOVA for repeated measures, N = 6-9 mice per group). (C) The efficacy of the pool of antagonists was validated against hyperalgesia evoked by PGE2 (9.8 nmol, intraplantarly) in naive C3H mice (2-way ANOVA for repeated measures (F[3, 80] = 8.88; P = .001, N = 5-6 mice per group). *Different from BL values at P < .013. #Different from PGE2 plus antagonists at P = .017; Bonferroni t test. (D) PGE2 increased cAMP in primary cultures of dissociated DRGs. The pool of EP receptor antagonists (10 μM per antagonist) blocked the effect of PGE2. Data are presented as picomoles of cAMP per well. *Different from other treatment groups and vehicle (V; ratio of DMSO 0.2% to ethanol 0.04% to saline 99.76%). Data were transformed to log10 for parametric statistical analysis. (F[3, 31] = 15.393; P < .001, 1-way ANOVA, N = 6 wells per treatment across 2 cultures).
PGE2-G produced hyperalgesia in HbAA-BERK mice. PGE2-G (2.3 nmol/10 µL, intraplantarly) was compared with the vehicle used for the pool of EP receptor antagonists (ratio of DMSO to ethanol to saline, 30%:1%:60%,10 µL, intraplantarly). (A) PGE2-G increased the frequency of withdrawal to a mechanical stimulus (3.9 mN). Coinjection of PGE2-G with vehicle or a pool of EP1, EP2, EP3, and EP4 receptor antagonists (SC51089, AH6809, L798106, and L161982, respectively, 20 nmol each, intraplantarly) did not reduce the effect of PGE2-G. Vehicle alone had no effect in HbAA-BERK mice. Two-way ANOVA for repeated measures demonstrated a significant treatment × time interaction (F[10, 106] = 2.67; P = .006, N = 8-9 mice per group). *Different from baseline (BL) and vehicle values at P < .004; Bonferroni t test. (B) The same pool of EP antagonists was ineffective in suppressing mechanical hyperalgesia in HbSS-BERK mice (F[1, 64] = 0.98; P = .326, 2-way ANOVA for repeated measures, N = 6-9 mice per group). (C) The efficacy of the pool of antagonists was validated against hyperalgesia evoked by PGE2 (9.8 nmol, intraplantarly) in naive C3H mice (2-way ANOVA for repeated measures (F[3, 80] = 8.88; P = .001, N = 5-6 mice per group). *Different from BL values at P < .013. #Different from PGE2 plus antagonists at P = .017; Bonferroni t test. (D) PGE2 increased cAMP in primary cultures of dissociated DRGs. The pool of EP receptor antagonists (10 μM per antagonist) blocked the effect of PGE2. Data are presented as picomoles of cAMP per well. *Different from other treatment groups and vehicle (V; ratio of DMSO 0.2% to ethanol 0.04% to saline 99.76%). Data were transformed to log10 for parametric statistical analysis. (F[3, 31] = 15.393; P < .001, 1-way ANOVA, N = 6 wells per treatment across 2 cultures).
An electrophysiological study confirmed that PGE2-G sensitized C-fiber nociceptors. Intraplantar injection of PGE2-G, but not vehicle, evoked activity, decreased mechanical threshold, and increased responses evoked by the suprathreshold mechanical stimulus in HbAA control mice (Figure 5A-D). Reduced thresholds to heat (Figure 5E) and cold stimuli (Figure 5F-G) following intraplantar injection of PGE2-G are additional evidence that PGE2-G sensitizes nociceptors. Although vehicle controls were not included in the assessment of temperature stimuli, the sensitization that was observed is not likely due to repeated administration of stimuli because thresholds remained constant using a similar stimulation protocol in naive mice.45
![Figure 5. PGE2-G sensitized C-fiber nociceptors in HbAA-BERK control mice. PGE2-G (23 nmol/10 μL) was administered intraplantarly. (A) PGE2-G evoked activity in C-fiber nociceptors in HbAA-BERK control mice compared with vehicle injection (F[4, 68] = 11.4; P < .005, 2-way ANOVA for repeated measures, treatment × time, N = 12 for vehicle, N = 10 for PGE2-G). Different from vehicle at *P < .05, **P < .01, ***P < .005. Different from time 0 (preinjection) at #P < .05, ###P < .005, ####P < .001 (Bonferroni t test). (B) PGE2-G decreased the threshold for response to a mechanical stimulus compared with vehicle (F[1, 20] = 20.2; P < .001, 2-way ANOVA for repeated measures for treatment, N = 12 for vehicle, N = 10 for PGE2-G). Mechanical response thresholds (millinewtons) were converted to percent change scores to enable parametric analyses. Different from vehicle at *P < .05, ***P < .005. (C) PGE2-G also increased responses evoked by a 149-mN von Frey monofilament (F[4, 68] = 4.5; P < .005, 2-way ANOVA for repeated measures, treatment × time). Different from vehicle at *P < .05, **P < .01. Different from time 0 at ##P < .01, ###P < .005, ####P < .001. (D) Representative examples of raw data demonstrating conduction latencies characteristic of C-fibers as well as changes in spontaneous activity and response to the 149-mN stimulus following injection of PGE2. Top, conduction latencies: multiple traces were overlapped to show consistency. The left arrow indicates the onset of the stimulus; the right arrow indicates the action potential of the fiber of interest. Middle, raw traces of 2 C-fiber nociceptors before and after treatment with vehicle or PGE2-G showing spontaneous activity. Bottom, representative raw data demonstrating responses to a 149-mN stimulus applied for 5 seconds. (E) Intraplantar PGE2-G administration decreased the threshold response to heat (F[4, 16] = 14.8; P < .001, ANOVA for repeated measures, N = 5). Different from time 0 at *P < .05, **P < .01 (Fisher least significant difference). (F) Intraplantar PGE2-G administration increased cold thresholds (increased temperature for response) (F[4, 16] = 3.7; P < .05, ANOVA for repeated measures, N = 5). *Different from P < .05 vs time 0. (G) Raw traces of a C-fiber nociceptor that did not respond to cold during initial characterization, but responded to cold following PGE2-G administration.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/133/18/10.1182_blood-2018-11-884346/3/m_blood884346f5.png?Expires=1765889841&Signature=SsmMHfUS-TL7jQK~xqjZ~L-C-rcrdP25jX5NqgUSYu0quegaTCkk130-0GBE3DMehGcYdn~Xy2TU-0-rzmsXP-GCxCQAdOYv12DVFUaSOT6xZfaBZ0A3H-fwhG4DquzucfOXU2mgKvZdHgrDTF2TwgPghjDMRDMo4rS1z8XMm7-zFaDLWpE6YinTdKysm~NU2r~XnJ5oMc2YIqDD-KascFraGVZjQwTJfOviGN1~~0A2njKY-K~JQVTCBHpSdsceWbF4Rmmxh52kCbl~QwEctO9t4ZSeZzhxBts3m8JWNW8BpXdKIgoUcl5gq1SFWk5oo7VzrJtA0YHZ0qcV~Ejw8A__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
PGE2-G sensitized C-fiber nociceptors in HbAA-BERK control mice. PGE2-G (23 nmol/10 μL) was administered intraplantarly. (A) PGE2-G evoked activity in C-fiber nociceptors in HbAA-BERK control mice compared with vehicle injection (F[4, 68] = 11.4; P < .005, 2-way ANOVA for repeated measures, treatment × time, N = 12 for vehicle, N = 10 for PGE2-G). Different from vehicle at *P < .05, **P < .01, ***P < .005. Different from time 0 (preinjection) at #P < .05, ###P < .005, ####P < .001 (Bonferroni t test). (B) PGE2-G decreased the threshold for response to a mechanical stimulus compared with vehicle (F[1, 20] = 20.2; P < .001, 2-way ANOVA for repeated measures for treatment, N = 12 for vehicle, N = 10 for PGE2-G). Mechanical response thresholds (millinewtons) were converted to percent change scores to enable parametric analyses. Different from vehicle at *P < .05, ***P < .005. (C) PGE2-G also increased responses evoked by a 149-mN von Frey monofilament (F[4, 68] = 4.5; P < .005, 2-way ANOVA for repeated measures, treatment × time). Different from vehicle at *P < .05, **P < .01. Different from time 0 at ##P < .01, ###P < .005, ####P < .001. (D) Representative examples of raw data demonstrating conduction latencies characteristic of C-fibers as well as changes in spontaneous activity and response to the 149-mN stimulus following injection of PGE2. Top, conduction latencies: multiple traces were overlapped to show consistency. The left arrow indicates the onset of the stimulus; the right arrow indicates the action potential of the fiber of interest. Middle, raw traces of 2 C-fiber nociceptors before and after treatment with vehicle or PGE2-G showing spontaneous activity. Bottom, representative raw data demonstrating responses to a 149-mN stimulus applied for 5 seconds. (E) Intraplantar PGE2-G administration decreased the threshold response to heat (F[4, 16] = 14.8; P < .001, ANOVA for repeated measures, N = 5). Different from time 0 at *P < .05, **P < .01 (Fisher least significant difference). (F) Intraplantar PGE2-G administration increased cold thresholds (increased temperature for response) (F[4, 16] = 3.7; P < .05, ANOVA for repeated measures, N = 5). *Different from P < .05 vs time 0. (G) Raw traces of a C-fiber nociceptor that did not respond to cold during initial characterization, but responded to cold following PGE2-G administration.
PGE2-G sensitized C-fiber nociceptors in HbAA-BERK control mice. PGE2-G (23 nmol/10 μL) was administered intraplantarly. (A) PGE2-G evoked activity in C-fiber nociceptors in HbAA-BERK control mice compared with vehicle injection (F[4, 68] = 11.4; P < .005, 2-way ANOVA for repeated measures, treatment × time, N = 12 for vehicle, N = 10 for PGE2-G). Different from vehicle at *P < .05, **P < .01, ***P < .005. Different from time 0 (preinjection) at #P < .05, ###P < .005, ####P < .001 (Bonferroni t test). (B) PGE2-G decreased the threshold for response to a mechanical stimulus compared with vehicle (F[1, 20] = 20.2; P < .001, 2-way ANOVA for repeated measures for treatment, N = 12 for vehicle, N = 10 for PGE2-G). Mechanical response thresholds (millinewtons) were converted to percent change scores to enable parametric analyses. Different from vehicle at *P < .05, ***P < .005. (C) PGE2-G also increased responses evoked by a 149-mN von Frey monofilament (F[4, 68] = 4.5; P < .005, 2-way ANOVA for repeated measures, treatment × time). Different from vehicle at *P < .05, **P < .01. Different from time 0 at ##P < .01, ###P < .005, ####P < .001. (D) Representative examples of raw data demonstrating conduction latencies characteristic of C-fibers as well as changes in spontaneous activity and response to the 149-mN stimulus following injection of PGE2. Top, conduction latencies: multiple traces were overlapped to show consistency. The left arrow indicates the onset of the stimulus; the right arrow indicates the action potential of the fiber of interest. Middle, raw traces of 2 C-fiber nociceptors before and after treatment with vehicle or PGE2-G showing spontaneous activity. Bottom, representative raw data demonstrating responses to a 149-mN stimulus applied for 5 seconds. (E) Intraplantar PGE2-G administration decreased the threshold response to heat (F[4, 16] = 14.8; P < .001, ANOVA for repeated measures, N = 5). Different from time 0 at *P < .05, **P < .01 (Fisher least significant difference). (F) Intraplantar PGE2-G administration increased cold thresholds (increased temperature for response) (F[4, 16] = 3.7; P < .05, ANOVA for repeated measures, N = 5). *Different from P < .05 vs time 0. (G) Raw traces of a C-fiber nociceptor that did not respond to cold during initial characterization, but responded to cold following PGE2-G administration.
A P2Y6 receptor antagonist blocked hyperalgesia in HbSS-BERK mice
The P2Y6 nucleotide receptor was recently identified as a cellular receptor for PGE2-G.36 Therefore, we tested whether MRS2578, a P2Y6 receptor antagonist,37 would reduce mechanical hyperalgesia in HbSS-BERK mice and block the development of hyperalgesia produced by PGE2-G in naive mice. Systemic administration of MRS2578 normalized the mechanical threshold in HbSS-BERK mice to that of HbAA-BERK mice but had no effect in HbAA-BERK mice (Figure 6A). Intraplantar administration confirmed a peripheral site for the drug action. A dose of 10 nmol (intraplantarly) relieved mechanical hyperalgesia from 30 to 90 minutes postadministration in HbSS-BERK mice (Figure 6B); a lower dose (3 nmol) was effective only at 30 minutes postadministration (data not shown). The ability of MRS2578 to block behavioral effects of PGE2-G was confirmed in naive C3H/He mice. The mechanical threshold in these mice is comparable to that of HbAA-BERK mice (compare baseline thresholds in Figure 6B-C). Intraplantar injection of PGE2-G increased sensitivity to mechanical (Figure 6C) and heat (Figure 6D) stimuli in C3H/He mice; coinjection with MRS2578 blocked this effect. It is noteworthy that MRS2578 had no effect on the response to either stimulus when administered alone.
![Figure 6. The P2Y6 receptor mediated effects of PGE2-G in HbSS-BERK mice. (A) A single systemic injection of the P2Y6 receptor antagonist, MRS2578 (30 nmol/50 μL, intraperitoneally), reduced mechanical allodynia in HbSS-BERK mice over time (2-way ANOVA for repeated measures (F[5, 45] = 2.65; P = .035, N = 7 mice per group). *Different from baseline (BL) in HbSS-BERK mice at P ≤ .038. #Different from HbAA-BERK mice at P ≤ .013; Bonferroni t test. The dose of MRS2578 that suppressed allodynia in HbSS mice did not change mechanical threshold in HbAA mice (P = 1.0, N = 5, Bonferroni t test). Legend in panel A is common to panel B. (B) Intraplantar injection of MRS2578 (10 nmol/10 μL, intraplantarly) blocked mechanical allodynia in HbSS-BERK mice acutely (2-way ANOVA for repeated measures; F[4, 36] = 3.39; P = .019, N = 7 mice per group). *Different from BL in HbSS-BERK mice at P ≤ .005. #Different from HbAA-BERK mice at P ≤ .017; Bonferroni t test. MRS2578 had no effect in HbAA mice (P ≥ .60, N = 4, Bonferroni t test). (C) Intraplantar injection of PGE2-G (2.3 nmol/10 μL, intraplantarly) produced mechanical allodynia in C3H mice and this effect was blocked by coinjection of the P2Y6 receptor antagonist, MRS2578 (3 nmol); 2-way ANOVA for repeated measures (F[3, 84] = 56.59; P > .001). *Different from BL within PGE2-G and from vehicle (ratio of DMSO to ethanol to saline, 0.7%:5%:94.3%) at P ≤ .003, N = 8-6 mice per treatment. #Different from PGE2-G plus MRS2578 at P ≤ .007, N = 6 mice per treatment, Bonferroni t test. MRS2578 alone had no effect (P = 1.0, N = 5, Bonferroni t test). Legend in panel C is common to panel D. (D) PGE2-G (2.3 nmol/10 μL, intraplantarly) increased sensitivity to radiant heat in C3H mice as defined by a decrease in PWL. Coinjection of MRS2578 (3 nmol) suppressed heat hyperalgesia produced by PGE2-G (2-way ANOVA RM (F[3, 52] = 9.97; P = .001, N = 5-8 mice per treatment). *Different from BL within PGE2-G–treated group and from vehicle at P ≤ .009. #Different from PGE2-G plus MRS2578 treatment at P ≤ .035, N = 6, Bonferroni t test. MRS2578 alone had no effect (P = .69, N = 5, Bonferroni t test).](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/133/18/10.1182_blood-2018-11-884346/3/m_blood884346f6.png?Expires=1765889841&Signature=Emkrbl89cic7HPXoJYgv2jNRMqcFxHSyvB1eKsHpwyhoP79SgVqq~iiW1A51JgPFKAfOpAX6KkVi2TAeF5r0xfZMUFXPpeBsEX8Y1HrwYzidsk7zSFRimZOS0GRo0UuxbwfNxOvRXuaTeEvcj5hm5XRpBbniZVPpdQJS7zD8RTKrN7Xgk2er3hIfKsWVZ95Zp4GLjohAQl2Baf70574-TBaYNKqy6qnVELuCdsM5Yg0bmC3OuuL4LilqL3HlNI-YOHN3xWsg1xi-MFh0yZ1~Q4oM8Z-wMS9DeBV4vLCy8j64WyQZSKnhqXd21ahb~cktDuuaXTywd40KMC0FoD3iEw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
The P2Y6 receptor mediated effects of PGE2-G in HbSS-BERK mice. (A) A single systemic injection of the P2Y6 receptor antagonist, MRS2578 (30 nmol/50 μL, intraperitoneally), reduced mechanical allodynia in HbSS-BERK mice over time (2-way ANOVA for repeated measures (F[5, 45] = 2.65; P = .035, N = 7 mice per group). *Different from baseline (BL) in HbSS-BERK mice at P ≤ .038. #Different from HbAA-BERK mice at P ≤ .013; Bonferroni t test. The dose of MRS2578 that suppressed allodynia in HbSS mice did not change mechanical threshold in HbAA mice (P = 1.0, N = 5, Bonferroni t test). Legend in panel A is common to panel B. (B) Intraplantar injection of MRS2578 (10 nmol/10 μL, intraplantarly) blocked mechanical allodynia in HbSS-BERK mice acutely (2-way ANOVA for repeated measures; F[4, 36] = 3.39; P = .019, N = 7 mice per group). *Different from BL in HbSS-BERK mice at P ≤ .005. #Different from HbAA-BERK mice at P ≤ .017; Bonferroni t test. MRS2578 had no effect in HbAA mice (P ≥ .60, N = 4, Bonferroni t test). (C) Intraplantar injection of PGE2-G (2.3 nmol/10 μL, intraplantarly) produced mechanical allodynia in C3H mice and this effect was blocked by coinjection of the P2Y6 receptor antagonist, MRS2578 (3 nmol); 2-way ANOVA for repeated measures (F[3, 84] = 56.59; P > .001). *Different from BL within PGE2-G and from vehicle (ratio of DMSO to ethanol to saline, 0.7%:5%:94.3%) at P ≤ .003, N = 8-6 mice per treatment. #Different from PGE2-G plus MRS2578 at P ≤ .007, N = 6 mice per treatment, Bonferroni t test. MRS2578 alone had no effect (P = 1.0, N = 5, Bonferroni t test). Legend in panel C is common to panel D. (D) PGE2-G (2.3 nmol/10 μL, intraplantarly) increased sensitivity to radiant heat in C3H mice as defined by a decrease in PWL. Coinjection of MRS2578 (3 nmol) suppressed heat hyperalgesia produced by PGE2-G (2-way ANOVA RM (F[3, 52] = 9.97; P = .001, N = 5-8 mice per treatment). *Different from BL within PGE2-G–treated group and from vehicle at P ≤ .009. #Different from PGE2-G plus MRS2578 treatment at P ≤ .035, N = 6, Bonferroni t test. MRS2578 alone had no effect (P = .69, N = 5, Bonferroni t test).
The P2Y6 receptor mediated effects of PGE2-G in HbSS-BERK mice. (A) A single systemic injection of the P2Y6 receptor antagonist, MRS2578 (30 nmol/50 μL, intraperitoneally), reduced mechanical allodynia in HbSS-BERK mice over time (2-way ANOVA for repeated measures (F[5, 45] = 2.65; P = .035, N = 7 mice per group). *Different from baseline (BL) in HbSS-BERK mice at P ≤ .038. #Different from HbAA-BERK mice at P ≤ .013; Bonferroni t test. The dose of MRS2578 that suppressed allodynia in HbSS mice did not change mechanical threshold in HbAA mice (P = 1.0, N = 5, Bonferroni t test). Legend in panel A is common to panel B. (B) Intraplantar injection of MRS2578 (10 nmol/10 μL, intraplantarly) blocked mechanical allodynia in HbSS-BERK mice acutely (2-way ANOVA for repeated measures; F[4, 36] = 3.39; P = .019, N = 7 mice per group). *Different from BL in HbSS-BERK mice at P ≤ .005. #Different from HbAA-BERK mice at P ≤ .017; Bonferroni t test. MRS2578 had no effect in HbAA mice (P ≥ .60, N = 4, Bonferroni t test). (C) Intraplantar injection of PGE2-G (2.3 nmol/10 μL, intraplantarly) produced mechanical allodynia in C3H mice and this effect was blocked by coinjection of the P2Y6 receptor antagonist, MRS2578 (3 nmol); 2-way ANOVA for repeated measures (F[3, 84] = 56.59; P > .001). *Different from BL within PGE2-G and from vehicle (ratio of DMSO to ethanol to saline, 0.7%:5%:94.3%) at P ≤ .003, N = 8-6 mice per treatment. #Different from PGE2-G plus MRS2578 at P ≤ .007, N = 6 mice per treatment, Bonferroni t test. MRS2578 alone had no effect (P = 1.0, N = 5, Bonferroni t test). Legend in panel C is common to panel D. (D) PGE2-G (2.3 nmol/10 μL, intraplantarly) increased sensitivity to radiant heat in C3H mice as defined by a decrease in PWL. Coinjection of MRS2578 (3 nmol) suppressed heat hyperalgesia produced by PGE2-G (2-way ANOVA RM (F[3, 52] = 9.97; P = .001, N = 5-8 mice per treatment). *Different from BL within PGE2-G–treated group and from vehicle at P ≤ .009. #Different from PGE2-G plus MRS2578 treatment at P ≤ .035, N = 6, Bonferroni t test. MRS2578 alone had no effect (P = .69, N = 5, Bonferroni t test).
Discussion
Using a murine model of human SCD, we have demonstrated that production of PGE2-G is sufficient to account for the sensitization of nociceptors and hyperalgesia, including increased sensitivity to mechanical, heat, and cold stimuli, which are associated with the disease. Acute administration of PGE2-G produced hyperalgesia and sensitization of C-fiber nociceptors that were equivalent to the levels observed in HbSS-BERK mice, and the reduction of hyperalgesia following R-flurbiprofen was consistent with decreased sensitization of nociceptors. The hyperalgesic effect of PGE2-G was documented previously in a model of inflammation induced by carrageenan.35 PGE2-G is generated by the oxidation of the endocannabinoid 2-AG by COX-2. COX-2 is not constitutively expressed in the periphery or in DRGs, so the production of PGE2-G is a consequence of induction of COX-2 in SCD and is consistent with the inflammatory course of the disease. When production of PGE2-G in HbSS-BERK mice was blocked by R-flurbiprofen, or P2Y6 receptors (the putative target of PGE2-G) were blocked with the selective antagonist MRS2578,37 hyperalgesia and sensitization of nociceptors were inhibited (Figure 7). Importantly, neither R-flurbiprofen nor MRS2578 had a behavioral effect in HbAA-BERK or naive C3H/He mice, suggesting that this compound is specific for conditions of inflammatory hyperalgesia as occurs in SCD and is not analgesic. Therefore, in uncovering a novel mechanism underlying pain in SCD, we have identified 2 pharmacological targets for reducing pain in patients with SCD: production of PGE2-G and P2Y6 receptors. These targets may provide important alternatives to high doses of opioids used to treat pain in SCD.
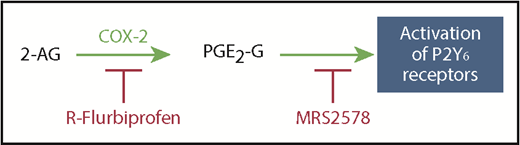
Schematic representation of the pathway for the generation of PGE2-G and its activation of P2Y6 receptors. COX-2 oxidizes 2-AG to generate PGE2-G, a putative agonist of P2Y6 receptors. This pathway is blocked by inhibition of COX-2 by R-flurbiprofen or blocking P2Y6 receptors with the antagonist MRS2578.
Schematic representation of the pathway for the generation of PGE2-G and its activation of P2Y6 receptors. COX-2 oxidizes 2-AG to generate PGE2-G, a putative agonist of P2Y6 receptors. This pathway is blocked by inhibition of COX-2 by R-flurbiprofen or blocking P2Y6 receptors with the antagonist MRS2578.
Although increased production of PGs is associated with vaso-occlusive crises in SCD,46 and NSAIDs are recommended to treat the associated pain,47 no controlled clinical trials to date have confirmed their efficacy in ameliorating pain in SCD6 and opioids are required for severe pain. Flurbiprofen is a slow reversible inhibitor of COX,48 and the R-enantiomer of flurbiprofen preferentially inhibits the oxidation of endocannabinoids over that of arachidonic acid, suggesting the possibility of substrate-selective inhibitors of COX-2.49 R-flurbiprofen also has an anti-inflammatory action through inhibition of NF-κB–dependent gene transcription50 that may provide additional benefit in SCD. Translation of R-flurbiprofen to use in humans will be facilitated by evidence that it was well tolerated in a clinical trial for treatment of Alzheimer disease.51 ARN2508 is an interesting new drug that combines the COX inhibitor flurbiprofen and the fatty acid amide hydrolase inhibitor URB597.52 The flurbiprofen portion of the drug retains its stereoselectivity, with R-ARN2508 also exhibiting selective inhibition of 2-AG oxidation compared with AA.53 Evidence that URB597 (the selective inhibitor of the enzyme fatty acid amide hydrolase, the primary enzyme that degrades the endocannabinoid anandamide) decreases nociceptor sensitization in HbSS-BERK sickle mice19 in conjunction with the present report suggests that R-ARN2508 may be especially suited for relief of pain in SCD. Furthermore, SCD may be a unique inflammatory disease. In contrast to the present results, S-flurbiprofen, but not the R-enantiomer, reduces central sensitization when COX-2 is increased in a rodent model of persistent inflammation.38
2-AG is the most abundant endocannabinoid, its levels exceeding those of anandamide by at least 50-fold.30,31 Within the central nervous system, 2-AG is an important mediator of analgesia,54,55 as well as in the periphery.56,57 Thus, a reduced level of 2-AG may contribute to hyperalgesia in SCD, and normalization of 2-AG following inhibition of its oxidation by COX-2 may contribute to the antihyperalgesic effect of R-flurbiprofen in sickle mice. Under physiological conditions, 2-AG is predominantly hydrolyzed by MGL.58 When COX-2 is elevated, as in chronic inflammation, 2-AG is oxygenized by COX-2 to form PGE2-G.28 As expected, this compound has distinct pharmacological properties from both endocannabinoids and PGs59 and mediates calcium mobilization, inositol 1,4,5-trisphosphate synthesis, and activation of protein kinase C in RAW264.7 macrophage cells.60 2-AG is synthesized by diacylglycerol lipase-β.61 Conversely, the present results are also consistent with a report that inhibition of this enzyme decreases production of 2-AG and therefore PGE2-G and produces a reduction in nociceptive behaviors in mice in a model of peripheral inflammation.62
Evidence that the P2Y6 receptor antagonist MRS2508 blocked the hyperalgesic effect of PGE2-G in naive C3H mice is consistent with a recent report that PGE2-G activates the purinergic receptor P2Y6.36 Moreover, PGE2-G mobilizes intracellular Ca2+ in vitro at ∼1 pM.60,63 Thus, although the level of PGE2-G measured in DRGs of HbSS-BERK mice was ∼0.6 fM, it is likely physiologically relevant. Furthermore, the absence of an effect of PGE2-G on the generation of cAMP is consistent with reports that PGE2-G does not activate prostanoid receptors.35,59,60 The cellular site of P2Y6 receptors that contributes to hyperalgesia in SCD remains to be resolved. In addition to expression by immune cells,64 P2Y6 receptors have been identified by western blot in DRGs and sciatic nerve,65 suggesting that they activate or sensitize nociceptors directly. Evidence that PGE2-G and MRS2508 had effects following intraplantar administration support the conclusion that immune cells and peripheral terminals of nociceptors contribute to pain in SCD.
In conclusion, we provide new information that PGE2-G, derived from the oxidation of the endocannabinoid 2-AG by COX-2 and through its interaction with the P2Y6 receptor, is a mediator of pain and nociceptor sensitization in SCD. R-flurbiprofen selectively blocked oxidation of 2-AG, reducing the production of PGE2-G and not PGE2, and normalizing the level of 2-AG. Inhibiting production of PGE2-G or blocking its putative target, P2Y6 receptors, reduced hyperalgesia and sensitization of nociceptors. The ability of R-flurbiprofen to block the synthesis of PGE2-G, and to normalize endocannabinoid tone66 suggests that R-flurbiprofen may be beneficial to treat pain in SCD when given alone or as an adjuvant to opioids, thereby lowering opioid doses needed to relieve pain and reducing opioid-related side effects.
For original data, please contact simon003@umn.edu.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors are grateful to P. Villalta in the Analytical Biochemistry Core facility of the University of Minnesota Masonic Cancer Center for direction in measurement of lipids, as well as to R. Jha and S. Kiven for assisting with breeding, phenotyping, and maintenance of sickle and control mice.
This work was supported by a grant from the National Institutes of Health, National Heart, Lung, and Blood Institute (R01 HL135895).
Authorship
Contribution: I.A.K. designed and executed biochemical and behavioral experiments, analyzed and interpreted data, and contributed to the writing of the manuscript; M.U. executed electrophysiological experiments, analyzed and interpreted data, and contributed to the writing of the manuscript; S.G.K. executed behavioral experiments and edited the manuscript; K.G. provided mice and edited the manuscript; and V.S.S. and D.A.S. contributed to the design of experiments, interpretation of data, and the writing of the manuscript.
Conflict-of-interest disclosure: K.G. is an advisor for Novartis, Tautona, Glycomimetics, and Fera Therapeutics, and has received a drug-only grant from Grifols; none of these have any conflict with the work presented in this manuscript. The remaining authors declare no competing financial interests.
The current affiliation for M.U. is Department of Anesthesia and Pain Medicine, University of Texas MD Anderson Cancer Center, Houston, TX.
Correspondence: Donald A. Simone, Department of Diagnostic & Biological Sciences, School of Dentistry, University of Minnesota, 17-252 Moos Tower, 515 Delaware St SE, Minneapolis, MN 55455; e-mail: simon003@umn.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal