

Key Points
Macrophages whose differentiation is skewed by VDR signaling are the key drivers for myofibroblasts in vivo.
Macrophages and VDR can be therapeutic targets in JAK2V617F-driven myelofibrosis.

Abstract
Myelofibrosis in myeloproliferative neoplasms (MPNs) with mutations such as JAK2V617F is an unfavorable sign for uncontrollable disease progression in the clinic and is complicated with osteosclerosis whose pathogenesis is largely unknown. Because several studies have revealed that macrophages are an indispensable supporter for bone-forming osteoblasts, we speculated that macrophages might play a significant role in the proliferation of collagen-producing myofibroblasts in marrow fibrotic tissues. Here, we show that myelofibrosis critically depends on macrophages whose differentiation is skewed by vitamin D receptor (VDR) signaling. In our novel myelofibrosis model established by transplantation of VDR+/+ hematopoietic stem/progenitor cells into VDR−/− mice, donor-derived F4/80+ macrophages proliferated together with recipient-derived α-smooth muscle actin–positive myofibroblasts, both of which comprised fibrotic tissues with an indistinguishable spindle-shaped morphology. Interfering VDR signals, such as low vitamin D diet and VDR deficiency in donor cells as well as macrophage depletion prevented myelofibrosis in this model. These interventions also ameliorated myelofibrosis in JAK2V617F-driven murine MPNs likely in a transforming growth factor-β1– or megakaryocyte-independent manner. These results suggest that VDR and macrophages may be novel therapeutic targets for MPNs with myelofibrosis.
Introduction
Philadelphia chromosome–negative myeloproliferative neoplasms (MPNs) include polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis (PMF). Nearly all patients with PV and more than half of patients with ET and PMF carry a somatic mutation of JAK2 V617F in hematopoietic cells.1,2 A common feature of MPNs is an initial hypercellular phase and, at later phase, fibrotic change in the bone marrow (BM), which is a predictive sign for the subsequent progression to massive splenomegaly and increased incidence of leukemic transformation.3 JAK inhibitors can reduce the size of an enlarged spleen but are not effective at reversing myelofibrosis and preventing leukemic transformation.4,5 This indicates the presence of unknown pathways that drive myelofibrosis in MPN patients other than constitutive JAK-STAT activation.
Myelofibrosis is characterized by the occupation of marrow space with spindle-shaped α-smooth muscle actin–positive (α-SMA+) stromal cells, so-called myofibroblasts, together with the accumulation of collagen fibers that can appear with silver staining.3 Recent studies have revealed that fibrosis-causing myofibroblasts were derived from their certain mesenchymal stromal precursors (Gli1+ and leptin receptor–positive [LepR+]) and a major driver for this proliferation/differentiation process is shown to be certain factors produced mainly by megakaryocytes such as transforming growth factor-β1 (TGF-β1) and platelet-derived growth factor (PDGF).6-8
A unique feature of myelofibrosis in MPNs is being a frequent companion of osteosclerosis,3 a thickening and irregularity of trabecular bone, whose pathogenesis is largely unknown. At the advanced stage of myelofibrosis, marrow space is replaced by the collagen tissues and trabecular bones, which suggests that myofibroblasts display an osteoblast-like function and myelofibrosis can be considered as a marrow ossification.
It has been shown that marrow macrophages are strong supporters for mesenchymal lineage cells.9-11 In particular, bone-associated OsteoMacs play important roles for the survival and activity of osteoblasts through at least, in part, the production of supportive factors such as oncostatin M and tumor necrosis factor-α.10-14 Depletion of macrophages including OsteoMacs results in subsequent disappearance of osteoblasts.11,12 Based on the critical function of macrophages for osteolineage-committed mesenchymal cells, we speculated that macrophages might play a significant role in the proliferation of collagen-producing myofibroblasts in the marrow fibrotic tissues.
Here, we show that myelofibrosis critically depends on macrophages whose differentiation is skewed by vitamin D receptor (VDR) signaling using myelofibrosis models of our original and JAK2V617F-driven MPNs.
Materials and methods
Mice and human BM biopsy samples
Mice were under the husbandry care of the Institute for Experimental Animals, Graduate School of Medicine, Kobe University. VDR−/− mice generated by gene targeting15 were backcrossed for >9 generations into a C57BL/6 background. Weanling mice (2-3 weeks old) were fed a high calcium diet to correct the major phenotype of VDR−/− mice: rickets type II represented by growth retardation and hypocalcemia16 (the composition of normal, high calcium [rescue diet for VDR−/− mice], and low vitamin D [high calcium] diet is shown in supplemental Table 1 [available on the Blood Web site]). C57BL/6-CD45.1 congenic mice and macrophage Fas-induced apoptosis (MaFIA) transgenic (Tg) mice17 were purchased from The Jackson Laboratory (Bar Harbor, ME). CAG-enhanced green fluorescent protein (EGFP) Tg mice were purchased from Japan SLC (Hamamatsu, Japan) and wild-type (WT) C57BL/6 mice were purchased from CLEA Japan (Chiba, Japan). JAK2V617F Tg mice were generated as described previously18 and backcrossed for >12 generations into a C57BL/6 background. Animals were maintained under specific pathogen-free conditions and on 12-hour light/12-hour darkness cycle and used for experiments at 6 to 8 weeks of age unless otherwise indicated. For the transplant recipients of JAK2V617F Tg or MaFIA/JAK2V617F double Tg donors, male C57BL/6 WT mice from CLEA Japan were used. Otherwise, both female and male mice were used in these studies. All animal studies were approved by the Animal Care and Use Committee of Kobe University.
BM biopsy samples of human MPN patients with or without severe myelofibrosis, which were collected for the purpose of diagnosis, were used for immunohistochemistry with written informed consent or opt-out approach under institutional review board approval at Kobe University (approval number 1455). Age, sex, and diagnosis were as follows: UPN1, 75 years old, female, PV; UPN2, 73 years old, male, PMF; UPN3, 72 years old, male, ET; UPN4, 75 years old, female, ET; UPN5, 54 years old, female, myelodysplastic syndrome/MPN; UPN6, 59 years old, male, myelodysplastic syndrome/MPN; UPN7, 63 years old, female, PV (under treatment with hydroxyurea); and UPN8, 78 years old, male, ET.
BM transplantation
Chimeric mice were generated by tail-vein injection of 2 × 106 CD45.1-WT donor BM nucleated cells (BMNCs) into lethally irradiated (14 Gy, 2 split doses with 3-hour interval) VDR+/+ or VDR−/− mice grown on a high calcium diet, among which VDR−/− recipients were referred to as the basic model of myelofibrosis. Blood was collected monthly to assess the blood cell counts and reconstitution by donor cells was confirmed by the CD45.1/CD45.2 chimerism of peripheral blood leukocytes. We also tested 2 × 106 BMNCs from VDR−/− mice (grown on a high calcium diet) and 1 × 105 CD45+lineage−c-kit+ cells sorted from WT CAG-EGFP Tg mice (grown on a normal diet) as donor cells. VDR+/+ and VDR−/− recipients were fed a high calcium diet even after transplantation.
BMNCs (5 × 106 cells) from VDR+/+, VDR+/+/JAK2V617F Tg (VDR+/+JAK), or VDR−/−/JAK2V617F Tg (VDR−/−JAK) mice (grown on a high calcium diet) were transplanted into WT (CD45.2) recipients (grown on a normal diet). Recipients were fed a normal diet also after transplantation. Blood was collected monthly to assess the blood cell counts, and organs such as BM and spleen were harvested 3 months after transplantation.
In vivo depletion of macrophages
CD45.1 BMNCs (1 × 107 cells) were transplanted into lethally irradiated (11 Gy [14 Gy was too harsh for the basic model mice with subsequent clodronate treatment], 2 split doses) VDR−/− recipient mice. Clodronate liposomes or vehicle (control liposomes) (clodronateliposome.org, Haarlem, The Netherlands) were injected intraperitoneally on posttransplantation day 3 (200 μL), day 5 (100 μL), and day 7 (100 μL) followed by 100 μL every 4 days up to 1 month when recipient femurs were harvested.
Lyophilized AP20187 (Apexbio, Houston, TX) was dissolved in dimethyl sulfoxide (Sigma-Aldrich, St. Louis, MO) at a concentration of 74 mg/mL and stored at −20°C before use. This stock solution was diluted with 10% PEG-400 and 1.7% Tween-20 in water just before intraperitoneal injection. BMNCs (1 × 107 cells) from MaFIA/JAK2V617F double Tg mice were injected into WT (CD45.2) recipients. Five weeks after transplantation, recipient mice were divided into 2 groups and the treatments of AP20187 or vehicle were started. Four sets of experiments were performed with different injection protocols because we noticed that some of these chimeric mice harboring MaFIA/JAK2V617F Tg BM lost their activity by the original administration schedule of AP20187 (10 mg/kg for 5 consecutive days followed by 1 mg/kg every 3 days).17 The actual doses in these experiments were as follows: first experiment, 10 mg/kg for 5 consecutive days followed by 1 mg/kg every 3.7 days on average (cumulative dose 1.14 mg); second experiment, 3 doses of 10 mg/kg every 2 days followed by 1 mg/kg every 5.6 days (cumulative dose 0.76 mg); third experiment, single dose of 10 mg/kg followed by 1 mg/kg every 4.0 days (cumulative dose 0.46 mg); and fourth experiment, single dose of 10 mg/kg followed by 1 mg/kg every 4.3 days (cumulative dose 0.43 mg). Three months after transplantation, blood, femur, and spleen were harvested. Similar results were obtained from all 4 experiments and pooled data are shown.
Statistical analysis
All data were pooled from at least 3 independent experiments. Immunohistochemistry and immunofluorescence staining were shown as representative data of 3 independent experiments that showed similar trends. All sample numbers (n) represent biological replicates. All center values shown in graphs refer to the mean. All values were reported as the mean plus or minus standard error of the mean (SEM). Statistical analysis was conducted using Kaplan-Meier analysis, the 2-tailed unpaired Student t test, the 1-way analysis of variance (ANOVA) test with Tukey post hoc procedure, and the Pearson correlation coefficient. No samples or animals were excluded from analysis, and sample size estimates were not used. Animals were randomly assigned to groups. Studies were not conducted blind with the exception of all histological analyses. Statistical significance was assessed with Prism (GraphPad Software, San Diego, CA) and defined as P < .05.
Complete methods for other procedures (cell lines, colony-forming unit in culture [CFU-C] assay, homing and engraftment, flow cytometry and cell sorting, micro–computed tomography [CT], tissue preparation and assessment of fibrosis, immunohistochemistry and immunofluorescence staining, assessment of macrophage and megakaryocyte numbers, bone histomorphometry, RNA extraction and quantitative real-time reverse transcription–polymerase chain reaction, and quantification of 1,25(OH)2D3 and intact parathyroid hormone [PTH]) are provided in supplemental Methods.
Results
Myelofibrosis develops in VDR−/− mice by transplantation of normal BM
During our study of impaired hematopoietic stem/progenitor cell (HSC/HPC) mobilization from the BM to circulation in VDR−/− mice,16 we found that BM of VDR−/− mice fed with a high calcium diet showed a significant increase of osteoblasts with no apparent hematopoietic alteration (supplemental Figures 1A-B, 2A-B, and 3A-B) and noticed that chimeric mice generated by transplantation of WT BM cells into lethally irradiated VDR−/− recipients (referred to as the basic model) showed lower recovery of platelet and white blood cell (WBC) counts compared with WT recipients, and died in 4 months (Figure 1A). Two months after transplantation in the femurs of the basic model, normal hematopoietic appearance was observed in the diaphysis. However, the BM cavity was occupied by monotonous spindle-shaped cells with a prominent increase of trabecular bones in the metaphysis (hematoxylin-eosin [H&E] staining in Figure 1B and CT in Figure 1C). Myelofibrosis was further confirmed by silver staining in VDR−/− recipient mice (Figure 1D). Osteoclast number as assessed by tartrate-resistant acid phosphatase staining was not decreased in the fibrotic area (data not shown). Megakaryocytes were not found in the fibrotic area and they were not increased in the hematopoietic area of diaphysis (data not shown). TGF-β in the blood was undetectable by enzyme-linked immunosorbent assay (data not shown). Although hematopoietic recovery seemed unaltered according to the appearance of diaphysis marrow cavity at 2 months after transplantation, a mild reduction of marrow cellularity and a drastic decline of hematopoietic progenitors (as assessed by CFU-Cs) in the BM were observed (supplemental Figure 4A). Because myelofibrosis in the metaphysis in the basic model was already observed at 1 month after transplantation (supplemental Figure 4B), and these chimeric mice died gradually in 4 months possibly due to insufficient hematopoiesis (Figure 1A), we speculated that this might be due to the loss of HSCs in association with myelofibrosis in the early phase after transplantation. The homing efficiency of HPCs (CFU-Cs and cell line FDCP mix) and HSCs (repopulating units assessed by serial transplantation) into the BM within a few hours after IV transplantation was normal (supplemental Figure 4C); however, HSC activities (repopulating units) were markedly reduced at 21 days after transplantation (supplemental Figure 4D). In other words, myelofibrosis in this basic model was associated with the loss of HSCs.
![Figure 1. Severe myelofibrosis and osteosclerosis develop in VDR−/− mice transplanted with VDR+/+ BM (basic model). (A) Survival and blood analyses (donor chimerism, WBC count, hemoglobin [Hb], and platelet count) in VDR+/+ and VDR−/− recipient mice transplanted with VDR+/+ BM. (B-D) Femur sections of VDR+/+ and VDR−/− recipient mice transplanted with VDR+/+ BM (2 months after transplantation): (B) H&E staining, (C) micro-CT, and (D) silver staining. Scale bars, 500 μm (black) and 50 μm (white). Representative pictures or combined data of at least 3 independent experiments are shown. Data are represented as mean plus or minus SEM. **P < .01, ***P < .001 (Kaplan-Meier analysis and Student t test).](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/133/15/10.1182_blood-2018-09-876615/3/m_blood876615f1.png?Expires=1765950001&Signature=4K2zwz5rvzdyf-jZ2iWmxqBYawKTAiSZT9siCyd7lZbOaYE5UX-qpHlxlYUqdVEUSLeWI0NeiQNlmORQkdK9TQ~dMDtEyvvfftQKH5O~mxr-xbAT0c8ggtBv29iPPtkSfcYJuPLLQo6AovIWmw6qmGWtKzXPmezs-ojTe7Vx-hJLeU0DZVFBUoX6B~cLh4TbrV66BDLPoV~4xQNUOSCzojzEPTCQCp7iYWVVrbMQrrdGx03kltlODUFwsr0UAXCUjcsQwJMNoyZuYpCTFIjhkZFFbFv6~ztsAg1sDcrbhhdVh8hjK7D9UL~4nrX5Y5XZri7g6NGSdsIbmGjJG0B7~w__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Severe myelofibrosis and osteosclerosis develop in VDR−/− mice transplanted with VDR+/+ BM (basic model). (A) Survival and blood analyses (donor chimerism, WBC count, hemoglobin [Hb], and platelet count) in VDR+/+ and VDR−/− recipient mice transplanted with VDR+/+ BM. (B-D) Femur sections of VDR+/+ and VDR−/− recipient mice transplanted with VDR+/+ BM (2 months after transplantation): (B) H&E staining, (C) micro-CT, and (D) silver staining. Scale bars, 500 μm (black) and 50 μm (white). Representative pictures or combined data of at least 3 independent experiments are shown. Data are represented as mean plus or minus SEM. **P < .01, ***P < .001 (Kaplan-Meier analysis and Student t test).
Severe myelofibrosis and osteosclerosis develop in VDR−/− mice transplanted with VDR+/+ BM (basic model). (A) Survival and blood analyses (donor chimerism, WBC count, hemoglobin [Hb], and platelet count) in VDR+/+ and VDR−/− recipient mice transplanted with VDR+/+ BM. (B-D) Femur sections of VDR+/+ and VDR−/− recipient mice transplanted with VDR+/+ BM (2 months after transplantation): (B) H&E staining, (C) micro-CT, and (D) silver staining. Scale bars, 500 μm (black) and 50 μm (white). Representative pictures or combined data of at least 3 independent experiments are shown. Data are represented as mean plus or minus SEM. **P < .01, ***P < .001 (Kaplan-Meier analysis and Student t test).
The blood level of PTH, as well as the active form of vitamin D [1,25(OH)2D3], is extremely high in VDR−/− recipients15 (supplemental Table 2), and it is well known that secondary hyperparathyroidism is associated with marrow fibrosis in humans.19 However, it was not the case in our model because chimeric mice generated by transplantation of VDR−/− BM cells into VDR−/− recipients survived normally (Figure 2A) and showed no myelofibrosis/osteosclerosis (Figure 2B) with normal blood cell counts (Figure 2C), suggesting that myelofibrosis in the basic model was critically associated with VDR signaling in hematopoietic cells. In addition to femoral marrow, similar fibrosis was also observed in the trabecular BM of the spine (supplemental Figure 5) and ribs (data not shown). All of these areas represent specific sites for immature hematopoietic cells to locate and initiate hematopoietic repopulation after transplantation.20 Collectively, our data suggest that transplanted VDR+/+ HSCs achieved trabecular BM in the mouse with high 1,25(OH)2D3 level and vanished after triggering the development of myelofibrosis/osteosclerosis.

Myelofibrosis and osteosclerosis do not develop in VDR−/− mice transplanted with VDR−/− BM. (A-C) Survival (A), H&E staining of femur sections (4 months after transplantation) (B), and blood cell counts (C) in VDR+/+ and VDR−/− recipient mice transplanted with VDR−/− BM. Scale bars, 500 μm (black) and 100 μm (white). Representative pictures or combined data of at least 3 independent experiments are shown. Data are represented as mean plus or minus SEM. *P < .05 (Kaplan-Meier analysis and Student t test).
Myelofibrosis and osteosclerosis do not develop in VDR−/− mice transplanted with VDR−/− BM. (A-C) Survival (A), H&E staining of femur sections (4 months after transplantation) (B), and blood cell counts (C) in VDR+/+ and VDR−/− recipient mice transplanted with VDR−/− BM. Scale bars, 500 μm (black) and 100 μm (white). Representative pictures or combined data of at least 3 independent experiments are shown. Data are represented as mean plus or minus SEM. *P < .05 (Kaplan-Meier analysis and Student t test).
Fibrotic tissues were composed of donor-derived macrophages and recipient-derived myofibroblasts
To confirm that this fibrotic tissue formation in VDR−/− recipients transplanted with WT BM cells was initiated by donor hematopoietic cells, we transplanted CD45+lineage−c-kit+ HSCs/HPCs sorted from WT CAG-EGFP Tg mice into lethally irradiated VDR−/− mice. We found that in this setting, VDR−/− recipients also developed myelofibrosis with osteosclerosis in the trabecular area of the femurs (Figure 3A). Thus, VDR+/+ immature hematopoietic cells are sufficient to initiate this phenomenon in a high 1,25(OH)2D3 microenvironment. In the metaphysis, monotonous spindle-shaped cells proliferated and many of these cells were GFP+ (Figure 3B). However, careful assessment revealed that these cells were composed of 2 distinct populations, F4/80+ macrophages and osterix+ mesenchymal cells with a strong expression of α-SMA, a marker for myofibroblasts (Figure 3C; supplemental Figure 6). These 2 populations were clearly distinguished by GFP or osterix expression. Macrophages were derived from donor cells (hematopoietic lineage, GFP+ in cytoplasm) and myofibroblasts were derived from VDR−/− host cells (red in nuclei [osterix+] and GFP−; Figure 3D). We also evaluated VDR messenger RNA (mRNA) expression in sorted immature hematopoietic cells by quantitative real-time reverse transcription–polymerase chain reaction and found that VDR mRNA in the lineage−c-kit+ immature fraction was selectively high compared with lineage-positive committed cells (Figure 3E). This suggested that, among hematopoietic cells, HSCs/HPCs might be selectively responsive to the high level of vitamin D. In line with this, 10 nM (4166 pg/mL) 1,25(OH)2D3, which is approximately equivalent to the level observed in the blood of the basic model (supplemental Table 2), was enough to skew the differentiation of the progenitor cell line HL-60 toward macrophage lineage in vitro (supplemental Figure 7A-B). To specify the major producers of collagen fibers, we assessed the expression of a collagen-specific molecular chaperone HSP47. HSP47 was positive in GFP− spindle-shaped cells (presumably myofibroblats) and bone surface–lining osteoblasts in the fibrotic tissue (supplemental Figure 8A-B). To test the possibility that these populations may contribute to MPNs, we also assessed immunohistochemical staining for macrophages and osteolineage cells in the BM of JAK2V617F Tg mice18 and the BM biopsy samples of MPN patients with severe fibrosis. The BM fibrotic tissue of JAK2V617F Tg mice was composed of CD169+ macrophages and osterix+ stromal cells (supplemental Figure 9). HSP47 was highly expressed in the osteoblasts lining the surface of the bone tissue as well as in the spindle-shaped cells in the fibrotic marrow area (supplemental Figure 9). As shown in supplemental Figure 10, human marrow fibrotic tissues also comprised both CD163+ macrophages and runx2+ stromal cells that were positive for α-SMA.

Marrow fibrotic tissue is composed of 2 distinct populations with identical morphology. (A-B) Femur sections of VDR+/+ and VDR−/− recipient mice transplanted with VDR+/+GFP+CD45+lineage−c-kit+ BM cells (2 months after transplantation): (A) H&E and silver staining and (B) GFP fluorescence. Scale bars, 500 μm (black) and 50 μm (white). (C-D) Immunohistochemical staining of F4/80, osterix, and α-SMA (C) and GFP expression and immunofluorescence staining of osterix (D) in marrow fibrotic tissue of VDR−/− recipient. Scale bars, 20 μm (black) and 20 μm (white). (E) VDR mRNA expression in lineage-positive (Lin+), Lin−c-kit+Sca1−, and Lin−c-kit+Sca1+ fractions from VDR+/+ BM (n = 3). Representative pictures or combined data of at least 3 independent experiments are shown. Data are represented as mean plus or minus SEM. **P < .01, ***P < .001 (ANOVA).
Marrow fibrotic tissue is composed of 2 distinct populations with identical morphology. (A-B) Femur sections of VDR+/+ and VDR−/− recipient mice transplanted with VDR+/+GFP+CD45+lineage−c-kit+ BM cells (2 months after transplantation): (A) H&E and silver staining and (B) GFP fluorescence. Scale bars, 500 μm (black) and 50 μm (white). (C-D) Immunohistochemical staining of F4/80, osterix, and α-SMA (C) and GFP expression and immunofluorescence staining of osterix (D) in marrow fibrotic tissue of VDR−/− recipient. Scale bars, 20 μm (black) and 20 μm (white). (E) VDR mRNA expression in lineage-positive (Lin+), Lin−c-kit+Sca1−, and Lin−c-kit+Sca1+ fractions from VDR+/+ BM (n = 3). Representative pictures or combined data of at least 3 independent experiments are shown. Data are represented as mean plus or minus SEM. **P < .01, ***P < .001 (ANOVA).
VDR and macrophages are therapeutic targets for myelofibrosis in the basic model
Because macrophages were derived from VDR+/+ BM cells and myofibroblasts were deficient in VDR in this basic model, we hypothesized that macrophages whose differentiation was skewed by VDR signaling promoted myelofibrosis. To test this idea, we fed the basic model mice with a low vitamin D diet. Strikingly, a low vitamin D diet prevented the increase of 1,25(OH)2D3 plasma levels and myelofibrosis/osteosclerosis (Figure 4A; supplemental Table 2). In addition, no reduction of immature hematopoietic cells in the BM and mature hematopoietic cells in the peripheral blood was observed in the basic model (Figure 4B; supplemental Figure 11A-B). Because some VDR−/− recipient mice showed diarrhea and significant mortality (Figure 4B), the intestine of VDR−/− mice might have low tolerance to a low vitamin D diet.
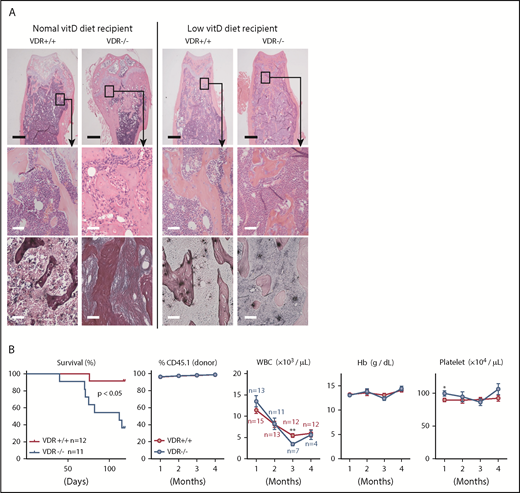
Low vitamin D diet prevents myelofibrosis and osteosclerosis in the basic model. (A) H&E and silver staining in femur sections of VDR+/+ and VDR−/− recipient mice transplanted with VDR+/+ BM (1 month after transplantation). Recipient mice were fed with a normal or low vitamin D diet after weaning, and transplanted at 7 to 8 weeks old. Scale bars, 500 μm (black) and 50 μm (white). (B) Survival, donor chimerism, and blood cell counts in VDR+/+ and VDR−/− recipient mice (low vitamin D diet) transplanted with VDR+/+ BM. Combined data of at least 3 independent experiments are shown. Data are represented as mean plus or minus SEM. *P < .05, **P < .01 (Kaplan-Meier analysis and Student t test).
Low vitamin D diet prevents myelofibrosis and osteosclerosis in the basic model. (A) H&E and silver staining in femur sections of VDR+/+ and VDR−/− recipient mice transplanted with VDR+/+ BM (1 month after transplantation). Recipient mice were fed with a normal or low vitamin D diet after weaning, and transplanted at 7 to 8 weeks old. Scale bars, 500 μm (black) and 50 μm (white). (B) Survival, donor chimerism, and blood cell counts in VDR+/+ and VDR−/− recipient mice (low vitamin D diet) transplanted with VDR+/+ BM. Combined data of at least 3 independent experiments are shown. Data are represented as mean plus or minus SEM. *P < .05, **P < .01 (Kaplan-Meier analysis and Student t test).
We next depleted macrophages by injection of clodronate liposome (supplemental Figure 12A-B). This treatment did not reduce the number of megakaryocytes (supplemental Figure 12C-D). Although osteosclerosis was not effectively rescued by this method, probably due to the effect as a bisphosphonate, myelofibrosis was clearly prevented (Figure 5A-B). Thus, VDR signaling in immature hematopoietic cells and subsequent differentiation/proliferation of macrophages are required for the progression of myelofibrosis (Figure 5C).

Depletion of macrophages prevents myelofibrosis in the basic model. (A-B) Silver and H&E staining in femur sections of VDR−/− recipient mice transplanted with VDR+/+ BM that were treated with clodronate liposome (1 month after transplantation). Silver staining of the metaphysis was digitized to distinguish fibrosis (blue), trabecular bone (red), and hematopoietic area (yellow; A) and enumerated as the ratio of each area (B; n = 6-8). Scale bars, 500 μm (black) and 50 μm (white). (C) Proposed concept of the basic model. Differentiation of immature hematopoietic cells is skewed toward macrophages, which likely drives myofibroblasts (and also osteoblasts presumably) as collagen producers, leading to myelofibrosis and osteosclerosis. Morphological marrow fibroblasts are composed of both macrophages and myofibroblasts. Representative pictures or combined data of at least 3 independent experiments are shown. Data are represented as mean plus or minus SEM. ***P < .001 (ANOVA).
Depletion of macrophages prevents myelofibrosis in the basic model. (A-B) Silver and H&E staining in femur sections of VDR−/− recipient mice transplanted with VDR+/+ BM that were treated with clodronate liposome (1 month after transplantation). Silver staining of the metaphysis was digitized to distinguish fibrosis (blue), trabecular bone (red), and hematopoietic area (yellow; A) and enumerated as the ratio of each area (B; n = 6-8). Scale bars, 500 μm (black) and 50 μm (white). (C) Proposed concept of the basic model. Differentiation of immature hematopoietic cells is skewed toward macrophages, which likely drives myofibroblasts (and also osteoblasts presumably) as collagen producers, leading to myelofibrosis and osteosclerosis. Morphological marrow fibroblasts are composed of both macrophages and myofibroblasts. Representative pictures or combined data of at least 3 independent experiments are shown. Data are represented as mean plus or minus SEM. ***P < .001 (ANOVA).
VDR and macrophages are therapeutic targets for JAK2V617F-driven myelofibrosis
Next, we tried to test the hypothesis that macrophages and VDR signaling could also play significant roles in myelofibrosis in a mouse model of JAK2V617F-driven MPNs. We bred JAK2V617F Tg mice with a low vitamin D diet after weaning. These mice did not show diarrhea or significant mortality. Although no improvement of blood cell counts was observed (Figure 6A), the BM of 5-month-old JAK2V617F Tg mice showed a significant reduction in myelofibrosis when fed with a low vitamin D diet, which led to a low plasma 1,25(OH)2D3 level (Figure 6B-D). More variation was observed in the degree of osteosclerosis, but overall it was not significant (Figure 6D).
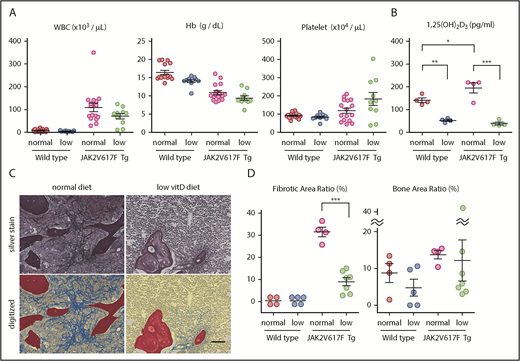
Low vitamin D diet prevents myelofibrosis in JAK2V617F Tg mice. (A-D) Blood cell counts (A; n = 9-15), plasma 1,25(OH)2D3 levels (B; n = 4), and silver staining in femur sections (C) of JAK2V617F Tg mice at 22 weeks old that were fed with a normal or low vitamin D diet after weaning. Pictures of the metaphysis were digitized to distinguish fibrosis (blue), trabecular bone (red), and hematopoietic area (yellow; C) and enumerated as the ratio of each area (D; n = 4-7). Scale bar, 50 μm. Representative pictures or combined data of at least 3 independent experiments are shown. Data are represented as mean plus or minus SEM. *P < .05, **P < .01, ***P < .001 (ANOVA).
Low vitamin D diet prevents myelofibrosis in JAK2V617F Tg mice. (A-D) Blood cell counts (A; n = 9-15), plasma 1,25(OH)2D3 levels (B; n = 4), and silver staining in femur sections (C) of JAK2V617F Tg mice at 22 weeks old that were fed with a normal or low vitamin D diet after weaning. Pictures of the metaphysis were digitized to distinguish fibrosis (blue), trabecular bone (red), and hematopoietic area (yellow; C) and enumerated as the ratio of each area (D; n = 4-7). Scale bar, 50 μm. Representative pictures or combined data of at least 3 independent experiments are shown. Data are represented as mean plus or minus SEM. *P < .05, **P < .01, ***P < .001 (ANOVA).
Next, to further assess the contribution of VDR signaling in MPN hematopoietic cells to myelofibrosis, we generated VDR+/+ and VDR−/− mice that harbored the JAK2V617F mutation (VDRJAK) by crossing both strains. Transplantation of VDR+/+JAK BM into lethally irradiated WT mice resulted in high WBC and platelet counts and a low hemoglobin level together with massive splenomegaly in 3 months, all of which were characteristic features of the late phase of MPN patients (Figure 7A; supplemental Figure 13A). These mice did not show diarrhea or significant mortality. In mice transplanted with VDR−/−JAK BM, alterations in blood cell counts showed a milder trend and the spleen size was significantly smaller compared with that of VDR+/+JAK BM-transplanted mice (Figure 7A; supplemental Figure 13A). Plasma 1,25(OH)2D3 levels were not significantly changed (supplemental Figure 13B). Silver staining of marrow 3 months after transplantation revealed severe myelofibrosis in VDR+/+JAK BM-transplanted WT mice, whereas myelofibrosis was reduced and sometimes absent in VDR−/−JAK BM-transplanted WT mice (Figure 7B-C; supplemental Figure 13C). As a significant contribution of TGF-β1 in myelofibrosis has been reported,21,22 we assessed TGF-β1 mRNA expression in the whole-bone tissue including BM. The averages were comparable between the 2 groups despite milder fibrosis in VDR−/−JAK BM-transplanted mice (Figure 7D). However, careful evaluation revealed a strong positive correlation between the severity of myelofibrosis and TGF-β1 mRNA level only in the absence of VDR (Figure 7D). These results suggest that, in addition to our basic model, VDR signaling in hematopoietic cells significantly contributes to JAK2V617F-driven myelofibrosis in a TGF-β1–independent manner.

VDR signaling and macrophages are therapeutic targets for JAK2V617F-driven myelofibrosis. (A-D) WT mice were transplanted with BM from WT (VDR+/+), VDR+/+/JAK2V617F Tg (VDR+/+JAK), or VDR−/−/JAK2V617F Tg (VDR−/−JAK) mice (n = 2, 9, and 10, respectively). Three months after transplantation, the appearance and size of the spleen (A; scale bars, 2 mm), silver staining of femur sections with enumeration of digitized pictures (B-C; scale bar, 50 μm), and the correlation between the severity of fibrosis and TGF-β1 mRNA expression in the bone tissue including BM (normalized to β-actin; D) were assessed. (E-G) WT mice transplanted with BM from JAK2V617F/MaFIA double Tg mice were treated with AP20187 to deplete macrophages. Three months after transplantation, silver staining of femur sections with enumeration of digitized pictures (E-F) and megakaryocyte numbers (G) were assessed (n = 4-7). Scale bar, 50 μm. Representative pictures or combined data of at least 3 independent experiments are shown. Data are represented as mean plus or minus SEM. **P < .01 (Student t test and Pearson correlation coefficient).
VDR signaling and macrophages are therapeutic targets for JAK2V617F-driven myelofibrosis. (A-D) WT mice were transplanted with BM from WT (VDR+/+), VDR+/+/JAK2V617F Tg (VDR+/+JAK), or VDR−/−/JAK2V617F Tg (VDR−/−JAK) mice (n = 2, 9, and 10, respectively). Three months after transplantation, the appearance and size of the spleen (A; scale bars, 2 mm), silver staining of femur sections with enumeration of digitized pictures (B-C; scale bar, 50 μm), and the correlation between the severity of fibrosis and TGF-β1 mRNA expression in the bone tissue including BM (normalized to β-actin; D) were assessed. (E-G) WT mice transplanted with BM from JAK2V617F/MaFIA double Tg mice were treated with AP20187 to deplete macrophages. Three months after transplantation, silver staining of femur sections with enumeration of digitized pictures (E-F) and megakaryocyte numbers (G) were assessed (n = 4-7). Scale bar, 50 μm. Representative pictures or combined data of at least 3 independent experiments are shown. Data are represented as mean plus or minus SEM. **P < .01 (Student t test and Pearson correlation coefficient).
Fibrotic marrow of VDR+/+JAK BM-transplanted mice contained both CD68+ monocytes/macrophages and runx2+ osteolineage cells (supplemental Figure 13D) and the numbers of macrophages and megakaryocytes in the marrow were comparable between VDR+/+JAK and VDR−/−JAK BM-transplanted mice (supplemental Figure 14A-B). To assess the role of macrophages in JAK2V617F-driven myelofibrosis, we used the MaFIA Tg mouse model, in which the CSF1 receptor promoter directs the expression of a ligand (AP20187)-inducible Fas-based suicide receptor in the mononuclear phagocyte lineage cells including OsteoMac, the bone-associated marrow macrophages.12,17 We generated JAK2V617F and MaFIA double Tg mice by crossing both strains and transplanted the BM into lethally irradiated WT mice. Depletion of macrophages by serial injection of AP20187 in chimeric mice that harbored JAK2V617F BM almost completely abrogated the development of myelofibrosis (Figure 7E-F; supplemental Figures 15 and 16A-B) with no reduction in BM megakaryocytes (Figure 7G; supplemental Figure 16C), blood cell counts, and spleen size (supplemental Figure 17). Collectively, macrophages whose differentiation is possibly skewed by VDR signaling largely regulate myelofibrosis induced by JAK2V617F mutation.
Discussion
Several models have been reported to explain the mechanism of myelofibrosis development. Based on the analyses of the effect of constitutive JAK-STAT activation in vivo, megakaryocyte-derived factors such as TGF-β1, PDGF, and CXCL4 are identified as essential stimulators for certain mesenchymal cells such as Gli1+ and Lepr+ stromal cells to develop myelofibrosis.6,8,21,22 In contrast, some reports have proposed that the key driver of myelofibrosis may be the myeloid lineage that harbors oncogenic mutations. Schepers et al reported that, using a mouse model of chronic myelogenous leukemia, chronic myelogenous leukemia myeloid cells stimulate multipotent stromal cells to overproduce functionally altered osteolineage cells, which leads to marrow fibrosis.23 Verstovsek et al showed the direct role of human neoplastic monocyte-derived fibrocytes in a xenograft model of PMF.24 Although mechanisms to achieve myelofibrosis vary depending on the models, our current study introduced macrophages as a novel important cell population to support the proliferation and activation of myofibroblasts in vivo.
The consensus for the definition of myofibroblasts would be (1) spindle-shaped morphology, (2) mesenchymal lineage (α-SMA+) cells, and (3) the production of collagen fibers. Osterix+ALCAM−Sca-1− preosteoblasts may fulfill these conditions. Because these cells are increased in the BM of VDR−/− mice (supplemental Figure 3), the VDR−/− microenvironment may have a potential for the hyperproliferation of myofibroblasts. However, according to our data that the transplantation of VDR−/− BM into VDR−/− recipients did not induce the proliferation of myofibroblasts (Figure 2), macrophages derived from VDR+/+ HSCs/HPCs are still indispensable for the formation of myelofibrosis in our basic model. It is not clear how macrophages drive the proliferation and activation of myofibroblasts. In addition to the supportive signals from macrophages to osteolineage cells through oncostatin M and tumor necrosis factor-α,13,14 it has also been reported that interleukin-1, which is produced by activated macrophages, enhances the response of osteolineage cells to PDGF,25 whereas TGF-β has been shown to induce the upregulation of macrophage colony-stimulating factor in osteolineage cells.26 Together with the pathological findings in fibrotic tissues of mice (Figure 3C-D) and humans (supplemental Figure 10), there may be a reciprocal amplification between macrophages and myofibroblasts through a complex relay of these growth factors. It is also possible that megakaryocytes contribute to this network by producing TGF-β and PDGF in MPNs.
Our basic model of myelofibrosis demonstrated that HSCs/HPCs with high-level expression of VDR mRNA differentiated in vivo into F4/80+ macrophages and HSCs/HPCs were lost from the BM with no extramedullary hematopoiesis. We have previously reported that 1,25(OH)2D3 induces the differentiation of hematopoietic progenitors toward macrophages in vitro.27 Based on our current study, this lineage skew of immature hematopoietic cells enforced by VDR signaling is likely a trigger for the activation of myofibroblasts as osteolineage cells and subsequent myelofibrosis/osteosclerosis. It was not clear whether this theory was applicable to human MPNs because the serum vitamin D level was reported to be not high in MPN patients.28 However, interruption of VDR signaling and macrophage depletion were clearly effective in preventing myelofibrosis of the JAK2V617F-driven MPN model. It is also possible that 1,25(OH)2D3 acts directly on differentiated macrophages because this axis has been shown to be important in some autoimmune diseases.29 Because macrophages express the enzyme responsible for the final hydroxylation step of 25-hydroxyvitamin D, the 1-α-hydroxylase30 and 1,25(OH)2D3 level in the blood was not drastically high in JAK2V617F Tg mice; macrophages could be a local producer of 1,25(OH)2D3 in fibrotic tissues in MPNs.
Megakaryocytes are not present in the fibrotic area in our basic model of myelofibrosis (supplemental Figure 12C-D), which suggests that they are not critical supporters for myofibroblasts depending on the model. In our study with a JAK2V617F-driven MPN model, a strong positive correlation between TGF-β1 mRNA expression and the severity of myelofibrosis appeared only in the absence of VDR in hematopoietic cells. Furthermore, the effect of macrophage ablation on the prevention of myelofibrosis was drastic despite no reduction of megakaryocytes. These indicate that, as a driver of myofibroblasts, macrophages are at least as important as megakaryocytes. It is also possible that the cross talk between megakaryocytes and macrophages might be important for myelofibrosis formation. Because VDR expression in megakaryocytes has been reported,31 vitamin D produced by macrophages may modulate megakaryocyte function in MPNs (a proposal in supplemental Figure 18).
We have shown in this study that myelofibrosis is a consequence of progressive deviation of interorgan communication between hematopoietic and skeletal systems. In the clinic, the development of efficient VDR antagonists and/or antimacrophage therapies may be promising strategies to control MPN patients with myelofibrosis.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Paul S. Frenette (Albert Einstein College of Medicine, New York) for the useful comments on the manuscript.
This work was supported by PRESTO, the Japan Science and Technology Agency (#JPMJPR12M7 [Y. Katayama]), a CREST grant from AMED (#JP18gm0910012h2 [Y. Katayama]), and the Grants-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (#15H04856, #18H02837 [Y. Katayama]) and for Scientific Research on Innovative Areas from the Ministry of Education, Culture, Sports, Science and Technology in Japan (#25118715 [Y. Katayama]). This work was also supported by the Daiichi Sankyo Foundation of Life Science, the Itochube Foundation, Novartis Pharma research grants, the Astellas Foundation for Research on Metabolic Disorders, and the SENSHIN Medical Research Foundation (Y. Katayama).
Authorship
Contribution: K.W. performed all experiments and wrote the manuscript; K.M., Y. Kawano, H.K., T.S., S.I., A.S., N.A., and M.S. helped with animal maintenance, tissue sample preparation, and capturing the images; S.K., K. Shide, K. Shimoda, and T.M. supervised the studies for VDR−/− and JAK2V617F Tg mice; and Y. Katayama supervised all experiments and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for K.M. is Hematology & Oncology Division, Penn State College of Medicine, Hershey, PA.
The current affiliation for Y. Kawano and H.K. is Endocrine/Metabolism Division, Wilmot Cancer Institute, University of Rochester Medical Center, Rochester, NY.
The current affiliation for N.A. is Department of Hematology and Oncology, Okayama University Hospital, Shikata-cho, Kita-ku, Okayama, Japan.
The current affiliation for M.S. is Oral Biochemistry and Molecular Biology, Graduate School of Dental Medicine, Hokkaido University, Sapporo, Japan.
Correspondence: Yoshio Katayama, Hematology, Department of Medicine, Kobe University Graduate School of Medicine, 7-5-1 Kusunoki-cho, Chuo-ku, Kobe 650-0017, Japan; e-mail: katayama@med.kobe-u.ac.jp.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal