

Abstract

β-Thalassemia is a common, frequently debilitating, inherited anemia caused by HBB gene mutations that reduce or eliminate the expression of the β-globin subunit of adult hemoglobin (HbA, α2β2). Consequently, excess free α-globin forms toxic precipitates in red blood cells (RBCs) and their precursors, leading to ineffective erythropoiesis and hemolytic anemia. Previously, we showed that free α-globin is eliminated by protein quality-control pathways, including the ubiquitin-proteasome system and autophagy (Khandros et al., Blood 2012;119:5265). In β-thalassemic mice, disruption of the Unc-51-like autophagy activating kinase gene (Ulk1) increased α-globin precipitates and worsened the pathologies of β-thalassemia. Treatment of β-thalassemic mice with rapamycin to inhibit mTOR (an ULK1 inhibitor) reduced α-globin precipitates, lessened ineffective erythropoiesis, and increased the lifespan of circulating RBCs in an Ulk1-dependent fashion.
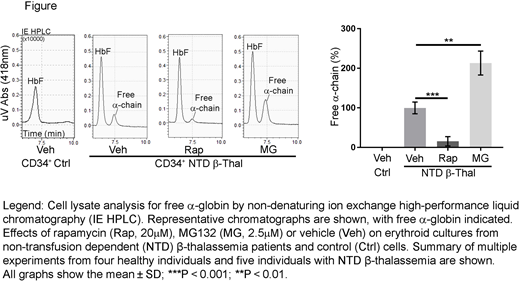
To investigate the therapeutic potential of rapamycin in human β-thalassemia, we treated erythroid precursors generated by in vitro differentiation of patient-derived CD34+ hematopoietic stem and progenitor cells. Reverse-phase high-performance liquid chromatography (HPLC) analysis of hemoglobinized erythroblasts generated from transfusion-dependent (TD, n = 5) or non-transfusion-dependent (NTD, n = 5) β-thalassemia patients revealed α-chain excesses (α-chain/β-like [β + γ + δ] chain) of approximately 40% and 15%, respectively (compared to 7 normal donors; P < 0.001). Rapamycin (10µM or 20µM) or the proteasome inhibitor MG132 (2.5µM) was added to day 13 cultures, which contained mid- to late-stage erythroblasts, and α-globin accumulation was determined by HPLC 2 days later. As expected, proteasome inhibition by MG132 raised free α-globin levels in thalassemic erythroblasts (P < 0.01) and induced cell death (P < 0.01). In contrast, rapamycin reduced free α-globin in a dose-dependent manner by 40% and 85% in TD (P < 0.0001) and NTD β-thalassemia (P < 0.001), respectively, but had no effect on erythroblasts derived from normal CD34+ cells (figure). We also observed decreases in the accumulation of autophagic markers, such as SQSTM1/p62 protein, by Western blotting. We observed no negative effects of rapamycin on the survival of patient-derived erythroblasts. Also of note, under our experimental conditions, rapamycin treatment of erythroblasts did not induce fetal hemoglobin production, as has been previously reported, thereby excluding this potential mechanism for reducing globin chain imbalances. Overall, rapamycin treatment significantly reduced the accumulation of free α-globin in TD β-thalassemia and almost fully corrected the imbalance in NTD β-thalassemia cells.
Our findings identify a new drug-regulatable pathway for ameliorating β-thalassemia. Rapamycin is approved and well studied, and it has a generally manageable toxicity profile. Moreover, there are additional pharmacologic approaches to activating ULK via mTOR inhibition or other pathways. These approaches may lead to effective drug therapies for β-thalassemia, particularly NTD or intermittently TD forms of the disease.
Cappellini:Celgene Corporation: Membership on an entity's Board of Directors or advisory committees; Vifor: Membership on an entity's Board of Directors or advisory committees; Sanofi/Genzyme: Membership on an entity's Board of Directors or advisory committees; Novartis: Honoraria.
This icon denotes a clinically relevant abstract

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal