

TO THE EDITOR:
Although the concept of somatic driver mutations in myeloproliferative neoplasms (MPNs) represented by polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis is well established,1-3 the contribution of germline or co-occurring JAK2 variants to a particular MPN phenotype is less well understood.4,5 Recently, 2 germline JAK2 mutations, E846D and R1063H, were described in a case of hereditary erythrocytosis6 ; the same JAK2 R1063H variant was initially reported in 3 of 93 PV patients who were JAK2 V617F+.7
In this study, we assessed the presence of JAK2 V617F and JAK2 R1063H mutations in a cohort of MPN patients to characterize the double-mutation carriers and gain insight into the functional consequences of coexisting mutations on JAK2 signaling.
Samples from 390 MPN patients positive for JAK2 V617F in Romania (n = 314) and Belgium (n = 76) were collected for the study. JAK2 R1063H mutation screening was performed using a custom TaqMan SNP Genotyping Assay. Fourteen of 390 JAK2 V617F+ MPN patients were found to carry concurrent JAK2 V617F and R1063H mutations. The clinical features and hematological data recorded at disease onset are summarized in supplemental Table 1 (available on the Blood Web site). ET was the most frequent diagnosis in double-mutation carriers (9/14). After considering bone marrow histology and the new World Health Organization 2016 criteria for PV diagnosis,8 2 patients were reconsidered to have PV. Our major finding is that a significantly higher white blood cell count (P = .023) and, correspondingly, a significantly higher neutrophil count (P = .025) were observed in double-mutation carriers compared with MPN patients harboring only the JAK2 V617F mutation (Figure 1A). When patients with an ET diagnosis were analyzed separately, we found that carriers of both mutations displayed a significantly higher neutrophil count (P = .031) and hemoglobin level (P = .046) compared with V617F+ ET patients (supplemental Figure 1). Furthermore, there was ≥1 thrombotic event during the course of the disease in 5 patients, including 2 cases of portal vein thrombosis. Interestingly, in a recent genomic study of patients with venous thromboembolism, JAK2 R1063H was identified in 1 case and was considered a probable disease-causing variant.9

Clinical characteristics, JAK2 analysis, and next-generation sequencing (NGS) screening for MPN patients exhibiting JAK2 V617F and JAK2 R1063H mutations. (A) Hematological data for JAK2 V617F MPN patients (n = 390) subdivided according to the JAK2 R1063H mutation status. Data for V617F only (n = 376) and for V617F/R1063H double-mutation carriers (n = 14) were recorded at diagnosis. For further information, see supplemental Material and methods and supplemental Table 1. The boxes represent the 25% to 75% interquartile range, horizontal lines within the boxes indicate medians, and vertical bars show the range of values (minimum to maximum). P values < .05 were considered statistically significant. *P < .05, Mann-Whitney U test. (B) JAK2 V617F allele burden, JAK2 R1063H fractional abundance, JAK2 V617F/R1063H mutations configuration, and additional mutations identified by targeted NGS. Third column: JAK2 V617F allele frequency obtained from the NGS study and compared with allele burden determined via quantitative PCR and ddPCR assay. Fourth column: JAK2 R1063H fractional abundance was determined using ddPCR in whole-blood samples collected at the time of diagnosis (see supplemental Material and methods for details). The JAK2 R1063H mutation was considered genuine germline only when the fractional abundance of the JAK2 R1063H variant was 50% (± 1.0%). Therefore, 8 patients are confirmed to be heterozygous germline carriers for the JAK2 R1063H variant. JAK2 R1063H in 3 samples with a percentage frequency of the mutant DNA between 20.7% and 31.5% (samples 1, 5, and 14) could be considered an acquired somatic mutation or an inherited variant that was partially lost due to UPD of the V617F–non-R1063H clone. Samples 4, 12, and 13 were nearly homozygous for JAK2 R1063H, and the presence of minor fraction of the WT allele excluded germline homozygosity. See also supplemental Figure 2. Fifth column: Cis/trans JAK2 V617F/R1063H mutations configuration was determined through sequencing of subcloned reverse-transcriptase PCR products spanning exons 14 through 24 of the JAK2 gene (see supplemental Material and methods for details). Sixth column: TruSight Myeloid Sequencing Panel (Illumina, San Diego, CA) was used for targeted mutational screening of JAK2 R1063H+ patients. Additional mutations were identified in 8 of 14 screened patients. A total of 11 variants was detected in 7 genes. #Five of these mutations are indexed in the Single Nucleotide Polymorphism database (dbSNP), and 4 of these specific variants are listed in the COSMIC catalog. One additional mutation in DNMT3A was published recently.14 §Two other mutations (frameshift in TET2 and premature stop codon in DNMT3A) do not have SNP/COSMIC IDs but are documented in the VarSome genomic variant database. **The GATA2 (A164T) allele (patient 12) was recently detected in a higher-than-expected frequency in myelodysplastic syndrome, suggesting a possible predisposing function in myeloid malignancies.15 Two patients harbor unique undescribed variants: patient 1 in BCOR and patient 9 in TET2. The BCOR variants were identified in 2 patients (both are missense mutations); their variant frequency was 54% for patient 1 and 99.5% for patient 11. They could be germline variants; both were estimated to be “damaging” or “probably damaging” by 2 algorithms (Sift, PolyPhen). *Indicates translation termination (stop) codon. All mutations were identified in DNA collected at the time of diagnosis; acquisition of additional mutations during disease evolution was not performed. For further information see supplemental Table 2. NA, not available; ND, not done; n.s., not significant; PMF, primary myelofibrosis; qPCR, quantitative PCR; snv, single nucleotide variant; Var Freq, variant allele frequency.
Clinical characteristics, JAK2 analysis, and next-generation sequencing (NGS) screening for MPN patients exhibiting JAK2 V617F and JAK2 R1063H mutations. (A) Hematological data for JAK2 V617F MPN patients (n = 390) subdivided according to the JAK2 R1063H mutation status. Data for V617F only (n = 376) and for V617F/R1063H double-mutation carriers (n = 14) were recorded at diagnosis. For further information, see supplemental Material and methods and supplemental Table 1. The boxes represent the 25% to 75% interquartile range, horizontal lines within the boxes indicate medians, and vertical bars show the range of values (minimum to maximum). P values < .05 were considered statistically significant. *P < .05, Mann-Whitney U test. (B) JAK2 V617F allele burden, JAK2 R1063H fractional abundance, JAK2 V617F/R1063H mutations configuration, and additional mutations identified by targeted NGS. Third column: JAK2 V617F allele frequency obtained from the NGS study and compared with allele burden determined via quantitative PCR and ddPCR assay. Fourth column: JAK2 R1063H fractional abundance was determined using ddPCR in whole-blood samples collected at the time of diagnosis (see supplemental Material and methods for details). The JAK2 R1063H mutation was considered genuine germline only when the fractional abundance of the JAK2 R1063H variant was 50% (± 1.0%). Therefore, 8 patients are confirmed to be heterozygous germline carriers for the JAK2 R1063H variant. JAK2 R1063H in 3 samples with a percentage frequency of the mutant DNA between 20.7% and 31.5% (samples 1, 5, and 14) could be considered an acquired somatic mutation or an inherited variant that was partially lost due to UPD of the V617F–non-R1063H clone. Samples 4, 12, and 13 were nearly homozygous for JAK2 R1063H, and the presence of minor fraction of the WT allele excluded germline homozygosity. See also supplemental Figure 2. Fifth column: Cis/trans JAK2 V617F/R1063H mutations configuration was determined through sequencing of subcloned reverse-transcriptase PCR products spanning exons 14 through 24 of the JAK2 gene (see supplemental Material and methods for details). Sixth column: TruSight Myeloid Sequencing Panel (Illumina, San Diego, CA) was used for targeted mutational screening of JAK2 R1063H+ patients. Additional mutations were identified in 8 of 14 screened patients. A total of 11 variants was detected in 7 genes. #Five of these mutations are indexed in the Single Nucleotide Polymorphism database (dbSNP), and 4 of these specific variants are listed in the COSMIC catalog. One additional mutation in DNMT3A was published recently.14 §Two other mutations (frameshift in TET2 and premature stop codon in DNMT3A) do not have SNP/COSMIC IDs but are documented in the VarSome genomic variant database. **The GATA2 (A164T) allele (patient 12) was recently detected in a higher-than-expected frequency in myelodysplastic syndrome, suggesting a possible predisposing function in myeloid malignancies.15 Two patients harbor unique undescribed variants: patient 1 in BCOR and patient 9 in TET2. The BCOR variants were identified in 2 patients (both are missense mutations); their variant frequency was 54% for patient 1 and 99.5% for patient 11. They could be germline variants; both were estimated to be “damaging” or “probably damaging” by 2 algorithms (Sift, PolyPhen). *Indicates translation termination (stop) codon. All mutations were identified in DNA collected at the time of diagnosis; acquisition of additional mutations during disease evolution was not performed. For further information see supplemental Table 2. NA, not available; ND, not done; n.s., not significant; PMF, primary myelofibrosis; qPCR, quantitative PCR; snv, single nucleotide variant; Var Freq, variant allele frequency.
To compare the frequency of additional somatic mutations in the analyzed patient groups, we used a targeted NGS panel for the 14 double-mutated patients and for 53 randomly selected patients from the JAK2 V617F cohort without the R1063H variant. Although a trend toward a higher mutational load was observed in the V617F/R1063H group compared with the V617F group, the difference did not reach statistical significance (P = .092) (supplemental Tables 2 and 3).
Next, we aimed to characterize the genotype and configuration of JAK2 mutations in the double-positive MPN patients. For these purposes, JAK2 V617F and R1063H allelic burdens were analyzed by quantitative polymerase chain reaction (PCR) and digital droplet PCR (ddPCR), and cis/trans configurations of JAK2 V617F and R1063H mutations were established by sequencing single colonies of subcloned JAK2 complementary DNA obtained from peripheral blood leukocytes in 10 of 14 double-mutated patients. Cis configuration of the mutations was detected in 6 cases, and trans configuration was found in 4 cases (Figure 1B). Quantification of the R1063H allele in the genomic DNA samples indicated that the variant was heterozygous in 8 cases, likely inherited, as shown previously6 (R1063H percentage ∼ 50%). In 3 patients with high V617F allelic burden, R1063H was nearly homozygous (fractional abundance > 80%), suggesting that 1 R1063H allele was inherited and the second allele was acquired by uniparental disomy (UPD). Low R1063H allele burden was found (allele percentage between 20.7% and 31.5%) in 3 other patients who exhibited trans configuration of JAK2 mutations (Figure 1B; supplemental Figure 2), raising the hypotheses that R1063H was acquired during the course of the disease or was partially lost due to UPD of the V617F–non-R1063H clone that, when amplified, decreased R1063H allelic burden. Because nonmyeloid tissue DNA was not available for the study, we used a combined array-comparative genomic hybridization/single-nucleotide polymorphism assay to detect unbalanced chromosomal changes and copy number neutral loss of heterozygosity in DNA samples with low R1063H allele burden. We detected UPD on chromosome 9p in 2 of 3 samples, suggesting that both hypotheses could be valid (supplemental Figure 3). However, without germline DNA, the origin of the R1063H mutation cannot be unequivocally established.
Later, cellular models were used to assess the functional consequences of the coexisting JAK2 mutations in cis or trans. Using site-directed mutagenesis, we generated JAK2 mutants (V617F, R1063H, and V617F/R1063H) on the background of JAK2 complementary DNA cloned into a pMEGIX–IRES–GFP bicistronic vector. STAT5 transcriptional activity of the JAK2 wild-type (WT) and JAK2 mutants in the presence of myeloid dimeric cytokine receptors (erythropoietin receptor [EPOR], thrombopoietin receptor [TPOR], and granulocyte-macrophage colony-stimulating factor receptor [G-CSFR]), as measured by dual-luciferase assay, revealed significantly higher constitutive activity of JAK2 V617F/R1063H (cis mutant) compared with JAK2 V617F in homozygous and heterozygous configurations and with each cytokine receptor (Figure 2A-C). Also, western blot analysis demonstrated a higher level of constitutive activation of JAK2 and STAT5 induced by the V617F/R1063H mutant vs V617F (Figure 2D). We then asked whether R1063H enhances the same conformational circuit used by V617F or triggers a different one. We introduced the E596R mutation in V617F/R1063H, because this mutation was previously found to block V617F constitutive, but not ligand-induced, JAK2 activity.10 The constitutive STAT5 transcriptional activities of V617F and V617F/R1063H mutants were decreased to the same extent by E596R (Figure 2E). Thus, R1063H amplifies signaling via the same circuit as V617F.
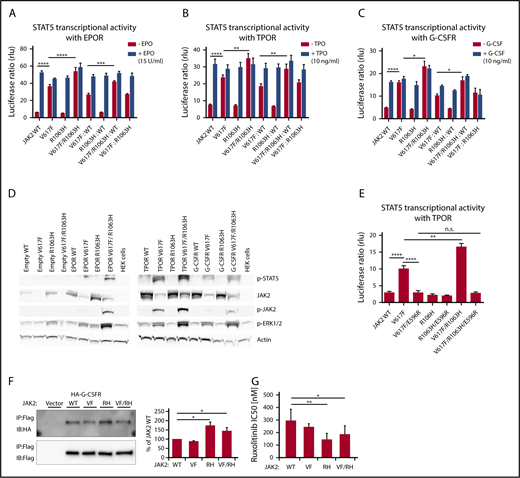
STAT5 transcriptional activity, the status of activation of downstream signaling by human JAK2 V617F and R1063H in the presence of dimeric myeloid cytokine receptors, the binding affinities of JAK2 mutants to G-CSFR and in vitro drug sensitivity assay. Constitutive and cytokine-dependent STAT5 transcriptional activity, as assessed by dual-luciferase assay, in γ2A cells transfected with JAK2 WT, JAK2 V617F, JAK2 R1063H, and JAK2 V617F/R1063H double mutant in the presence of EPOR (A), TPOR (B), and G-CSFR (C). Homozygous and heterozygous states of JAK2 mutants are mimicked. Shown are the averages of 9 replicates from 3 independent experiments ± standard error of the mean (SEM). *P < .05, **P < .01, ***P < .001, ****P < .0001, 1-way analysis of variance (ANOVA), followed by the post hoc Tukey test. (D) Western blot analysis of constitutive JAK2, STAT5, and ERK 1/2 phosphorylation levels (indicative of activated status) induced by human JAK2 mutants coexpressed with empty vector/cytokine receptors in HEK 293T cells. β-actin antibody was used as a loading control. Higher levels of p-JAK2 (p-Tyr1007/1008), p-STAT5 (p-Tyr694), and p-ERK 1/2 (p-Thr 202/p-Tyr 204) are observed in cells expressing the JAK2 V617F/R1063H double mutant in comparison with JAK2 V617F. Image shown is representative of 3 independent experiments. (E) The effect of E596R mutation on constitutive STAT5 activation induced by JAK2 V617F and JAK2 V617F/R1063H, as evaluated by dual-luciferase assay, in γ2A cells in the presence of TPOR. Both mutated proteins exhibit a similar decline in constitutive activity. The graph displays the averages of 9 replicates from 3 independent experiments ± SEM. **P < .01, ****P < .0001, 1-way ANOVA, followed by the post hoc Tukey test. (F) JAK2 mutants bind to the cytokine receptor G-CSFR with different affinities. Flag-tagged JAK2 mutants were transiently expressed in HEK 293 cells in which hemagglutinin-tagged G-CSFR was stably expressed. Interaction was examined by coimmunoprecipitation with anti-Flag affinity gel. Immunoblot band intensity was quantified by ImageJ software and normalized to a loading control, and WT JAK2 intensity was set to 100%. The data represent the mean of 3 independent experiments; T bars designate SEM. See also supplemental Figure 4. P values < .05 were considered statistically significant. *P < .05, Student paired t test with equal variance. (G) In vitro drug sensitivity assay. Stably transfected Ba/F3/EPOR cells expressing JAK2 WT, JAK2 V617F, JAK2 R1063H, and JAK2 V617F/R1063H were cultivated for 72 hours with decreasing concentrations of the JAK2 inhibitor ruxolitinib (1, 0.5, 0.25, 0.1, 0.05, 0.01, 0.001, and 0 µM). IC50 was defined as the drug concentration needed to inhibit 50% of cell growth (using GraphPad Prism 6.01 software). The data represent the mean of 7 independent experiments performed in triplicates (see also supplemental Material and methods for details). T bars designate the standard deviation. When the ruxolitinib sensitivity of mutant cells is compared with WT cells, the sensitivity of V617F+ cells is not statistically significant, whereas R1063H+ and V617F/R1063H double-mutant cells are significantly more sensitive to ruxolitinib compared with WT cells. Experiments with AZ-960 revealed comparable results (data not shown). P < .05 was considered statistically significant. *P < .05, **P < .01, 1-way ANOVA, followed by the post hoc Tukey test. IB, immunoblot; IP, immunoprecipitation; RH, R1063H; rlu, relative light unit; VF, V617F; VF/RH, V617F/R1063H.
STAT5 transcriptional activity, the status of activation of downstream signaling by human JAK2 V617F and R1063H in the presence of dimeric myeloid cytokine receptors, the binding affinities of JAK2 mutants to G-CSFR and in vitro drug sensitivity assay. Constitutive and cytokine-dependent STAT5 transcriptional activity, as assessed by dual-luciferase assay, in γ2A cells transfected with JAK2 WT, JAK2 V617F, JAK2 R1063H, and JAK2 V617F/R1063H double mutant in the presence of EPOR (A), TPOR (B), and G-CSFR (C). Homozygous and heterozygous states of JAK2 mutants are mimicked. Shown are the averages of 9 replicates from 3 independent experiments ± standard error of the mean (SEM). *P < .05, **P < .01, ***P < .001, ****P < .0001, 1-way analysis of variance (ANOVA), followed by the post hoc Tukey test. (D) Western blot analysis of constitutive JAK2, STAT5, and ERK 1/2 phosphorylation levels (indicative of activated status) induced by human JAK2 mutants coexpressed with empty vector/cytokine receptors in HEK 293T cells. β-actin antibody was used as a loading control. Higher levels of p-JAK2 (p-Tyr1007/1008), p-STAT5 (p-Tyr694), and p-ERK 1/2 (p-Thr 202/p-Tyr 204) are observed in cells expressing the JAK2 V617F/R1063H double mutant in comparison with JAK2 V617F. Image shown is representative of 3 independent experiments. (E) The effect of E596R mutation on constitutive STAT5 activation induced by JAK2 V617F and JAK2 V617F/R1063H, as evaluated by dual-luciferase assay, in γ2A cells in the presence of TPOR. Both mutated proteins exhibit a similar decline in constitutive activity. The graph displays the averages of 9 replicates from 3 independent experiments ± SEM. **P < .01, ****P < .0001, 1-way ANOVA, followed by the post hoc Tukey test. (F) JAK2 mutants bind to the cytokine receptor G-CSFR with different affinities. Flag-tagged JAK2 mutants were transiently expressed in HEK 293 cells in which hemagglutinin-tagged G-CSFR was stably expressed. Interaction was examined by coimmunoprecipitation with anti-Flag affinity gel. Immunoblot band intensity was quantified by ImageJ software and normalized to a loading control, and WT JAK2 intensity was set to 100%. The data represent the mean of 3 independent experiments; T bars designate SEM. See also supplemental Figure 4. P values < .05 were considered statistically significant. *P < .05, Student paired t test with equal variance. (G) In vitro drug sensitivity assay. Stably transfected Ba/F3/EPOR cells expressing JAK2 WT, JAK2 V617F, JAK2 R1063H, and JAK2 V617F/R1063H were cultivated for 72 hours with decreasing concentrations of the JAK2 inhibitor ruxolitinib (1, 0.5, 0.25, 0.1, 0.05, 0.01, 0.001, and 0 µM). IC50 was defined as the drug concentration needed to inhibit 50% of cell growth (using GraphPad Prism 6.01 software). The data represent the mean of 7 independent experiments performed in triplicates (see also supplemental Material and methods for details). T bars designate the standard deviation. When the ruxolitinib sensitivity of mutant cells is compared with WT cells, the sensitivity of V617F+ cells is not statistically significant, whereas R1063H+ and V617F/R1063H double-mutant cells are significantly more sensitive to ruxolitinib compared with WT cells. Experiments with AZ-960 revealed comparable results (data not shown). P < .05 was considered statistically significant. *P < .05, **P < .01, 1-way ANOVA, followed by the post hoc Tukey test. IB, immunoblot; IP, immunoprecipitation; RH, R1063H; rlu, relative light unit; VF, V617F; VF/RH, V617F/R1063H.
The enhancement of G-CSFR signaling, which regulates neutrophilic granulocyte formation, by V617F/R1063H might be relevant for the neutrophilia, which is not seen in all MPN patients with JAK2 V617F. Neutrophilia is detected in JAK2-double mutant patients, irrespective of cis- or trans-configuration. For the latter, we could not see enhanced activation by R1063H and V617F vs WT JAK2 and V617F via cytokine receptors (Figure 2A-C), possibly because small changes are difficult to detect in overexpression systems. We assessed whether R1063H changes the association of JAK2 with G-CSFR. Using coimmunoprecipitation, we detected a significantly higher biochemical association between JAK2 R1063H and JAK2 V617F/R1063H with G-CSFR compared with JAK2 V617F or JAK2 (Figure 2F). This might have a significant impact on signaling at low receptor levels in vivo, as well as in a trans-configuration, because R1063H alone enhances the association of JAK2 with G-CSFR. Linking neutrophilia to the increased association of JAK2 V617F/R1063H with G-CSFR is in agreement with a recent study in which differential coupling of JAK2 mutants to different receptors impacted the in vivo phenotypes induced by the different mutants.11 In ET double-mutant carriers, the higher level of hemoglobin that accompanied the higher neutrophil count supports the hypothesis that the co-occurrence of JAK2 V617F and R1063H mutations would lead to an ET phenotype with PV-like features, as a result of a cumulative effect on JAK2 signaling.12 Furthermore, the ruxolitinib sensitivity of JAK2 V617F/R1063H–expressing cells may have therapeutic implications (Figure 2G).
The frequency of R1063H in our JAK2 V617F+ MPN cohort (14/390) is consistent with the initial report (3/93 PV patients).7 The frequency of R1063H cited in the normal population in the Exome Aggregation Consortium database13 is much lower (0.004377). More studies on large patient populations, as well as on families with MPNs, would be necessary for determining whether JAK2 R1063H predisposes to acquisition of the JAK2 V617F mutation, as well as to assess its role in MPN progression, given the involvement of G-CSFR in leukemia.
The online version of this article contains a data supplement.
Acknowledgments
The authors gratefully acknowledge funding from the Competitiveness Operational Program A1.1.4. ID: P_37_798, contract 149/26.10.2016 (MySMIS2014+: 106774), Molecular Profiling of Myeloproliferative Neoplasms and Acute Leukemia Project. S.N.C. received support from the Ludwig Institute for Cancer Research, Fondation contre le cancer, Salus Sanguinis, Fondation Les Avions de Sébastien, the Action de Recherche Concertée Project, and the Walloon Excellence in Lifesciences & Biotechnology project, Belgium. O.B., B.K., L.L., and V.D. received research funding from the Czech Science Foundation (project GACR 17-05988S) and from the Ministry of Education, Youth and Sports, Czech Republic (projects LO1220 [O.B.] and LTAUSA17142 [B.K., L.L., and V.D.]). M.B. and J.V. received support from the Ministry of Health of Czech Republic– conceptual development of research organization (00023736).
Authorship
Contribution: C.M., O.B., and J.-P.D. designed and performed research, analyzed data, and wrote the manuscript; L.N., O.S., A.T., N.B., D.C., V.H., P.S., and C.C.D. recruited the patients, performed research, and contributed to the editing of the manuscript; E.L., B.K., M.B., J.V., L.L., and C.P. performed research, analyzed data, and reviewed the manuscript; and S.N.C. and V.D. designed the study, wrote the manuscript, and provided financial support.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Stefan N. Constantinescu, Ludwig Institute for Cancer Research, Ave Hippocrate 74, UCL, 75-4, 1200 Brussels, Belgium; e-mail: stefan.constantinescu@bru.licr.org; and Vladimir Divoky, Department of Biology, Faculty of Medicine and Dentistry, Palacky University, Hnevotinska 3, CZ-775 15 Olomouc, Czech Republic; e-mail: vladimir.divoky@upol.cz.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal