

Abstract
Abdominal aortic aneurysm (AAA) is a degenerative vascular pathology resulting in significant morbidity and mortality in older adults due to rupture and sudden death. Despite 150 000 new cases and nearly 15 000 deaths annually, the only approved treatment of AAA is surgical or endovascular intervention when the risk for aortic rupture is increased. The goal of the scientific community is to develop novel pharmaceutical treatment strategies to reduce the need for surgical intervention. Because most clinically relevant AAAs contain a complex structure of fibrin, inflammatory cells, platelets, and red blood cells in the aneurysmal sac known as an intraluminal thrombus (ILT), antithrombotic therapies have emerged as potential pharmaceutical agents for the treatment of AAA progression. However, the efficacy of these treatments has not been shown, and the effects of shrinking the ILT may be as detrimental as they are beneficial. This review discusses the prospect of anticoagulant and antiplatelet (termed collectively as antithrombotic) therapies in AAA. Herein, we discuss the role of the coagulation cascade and platelet activation in human and animal models of AAA, the composition of ILT in AAA, a possible role of the ILT in aneurysm stabilization, and the implications of antithrombotic drugs in AAA treatment.
Medscape Continuing Medical Education online
![]()
In support of improving patient care, this activity has been planned and implemented by Medscape, LLC and the American Society of Hematology. Medscape, LLC is jointly accredited by the Accreditation Council for Continuing Medical Education (ACCME), the Accreditation Council for Pharmacy Education (ACPE), and the American Nurses Credentialing Center (ANCC), to provide continuing education for the healthcare team.
Medscape, LLC designates this Journal-based CME activity for a maximum of 1.00 AMA PRA Category 1 Credit(s)™. Physicians should claim only the credit commensurate with the extent of their participation in the activity.
All other clinicians completing this activity will be issued a certificate of participation. To participate in this journal CME activity: (1) review the learning objectives and author disclosures; (2) study the education content; (3) take the post-test with a 75% minimum passing score and complete the evaluation at http://www.medscape.org/journal/blood; and (4) view/print certificate. For CME questions, see page 2701.
Disclosures
CME questions author Laurie Barclay, freelance writer and reviewer, Medscape, LLC, owns stock, stock options, or bonds from Pfizer. Associate Editor Thomas L. Ortel and the authors declare no competing financial interests.
Learning objectives
Upon completion of this activity, participants will be able to:
Describe the potential role and implications in abdominal aortic aneurysm of antiplatelet agents, based on a review
Describe the potential role and implications in abdominal aortic aneurysm of anticoagulants, based on a review
Describe other factors to consider in the decision to start antithrombotic therapy in patients with abdominal aortic aneurysm, based on a review
Release date: December 20, 2018; Expiration date: December 20, 2019
Introduction
An infrarenal abdominal aortic aneurysm (AAA) is a permanent, localized dilation in the aortic wall defined by a diameter 50% greater than an adjacent, presumably normal aortic segment (≥3.0 cm in adult patients).1,2 According to established guidelines, male patients 65 to 75 years of age who have smoked at least 100 cigarettes in their lifetime should have a 1-time screen for AAA. Unfortunately, many patients develop rapidly expanding AAA despite this recommendation. Static images are insufficient to assess true risk for AAA expansion and rupture, and greater attention should be paid to understanding the biological components of vascular integrity, which may predict aneurysmal growth.3 Morbidity and mortality from AAA rupture is the 13th leading cause of death in the United States, affecting 5% to 10% of men and 1% to 2% of women over the age of 65.2,4,5 Open surgical or percutaneous endovascular aneurysm repair (EVAR) intervention is the only treatment of AAA, pursued when the aneurysm has reached the “maximum diameter criterion” (>5.5 cm), rapid expansion (0.5 cm) in the preceding 6 months, or if the dissection flap compromises blood flow to aortic branch vessels (renal or iliac arteries), which may compromise end-organ perfusion.2,6,7 Patients with AAA constitute a diagnosis of peripheral vascular disease (PVD), and so treatment with low-dose aspirin (ASA) or a P2Y12 receptor antagonist for the purposes of secondary prevention is consistent with established guidelines because there are no validated pharmacological therapies for the prevention of AAA progression or rupture.2,8,9
Pathophysiologically, AAAs are characterized by a failure of the major structural proteins of the aorta (ie, elastin and collagen), resulting in proteolytic degradation of extracellular matrix, apoptosis of vascular smooth muscle cells (VSMCs), and eventual thinning of the aortic tunica media.10,11 Resulting from this loss of resistance in the medial aorta, outward centrifugal mass transport of soluble blood components through the wall increases via hydraulic conductance, resulting in inflammation, edema, and lymphoid neogenesis in the adventitia.12 In addition, the adventitia is a highly vascularized and innervated connective tissue whereby this accumulation of inflammatory cells is linked to capillary development and a staging site for macrophage and lymphocyte infiltration into the vascular media.10,12 This inflammatory response is associated with the formation of a nonocclusive mural thrombus, or intraluminal thrombus (ILT; Figure 1), involving the activation of both platelets and proteins within the coagulation cascade.2 Collectively, these processes lead to the widening of the vessel lumen, loss of structural integrity of the aorta, and an increased susceptibility to rupture and death.11
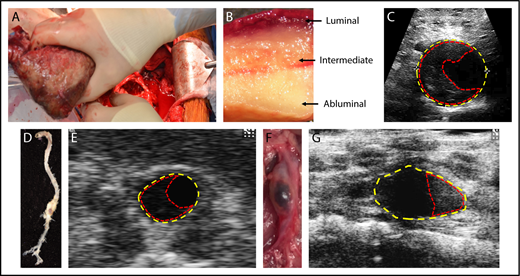
Composition of the ILT in humans and mouse models. (A) Representative picture of a 4.6-cm-diameter ILT (42.9 cm2) being removed from a 6-cm diameter aneurysm (79.4 cm2) during an open surgical procedure. (B) Macroscopic view of an ILT demonstrating the luminal, intermediate, and abluminal layers. (C) Ultrasound image of a human AAA with yellow and red dashed lines outlining the area of the aneurysm and thrombus, respectively. (D) A representative suprarenal AAA aorta from a low-density lipoprotein receptor–deficient (Ldlr−/−) mouse infused with AngII (1000 ng/kg per min) with an (E) in vivo ultrasound image captured at the maximum diameter on a Vevo 2100 (FUJIFILM VisualSonics Inc), where yellow dashed lines represent the area of the aneurysm and red dashed lines outline the extraluminal thrombus. (F) A representative infrarenal AAA aorta from a C57BL/6J mouse treated with topical elastase to induced aneurysm and administration of β3-aminopropionitrile fumarate salt (BAPN) ad libitum via water bottle to induce an ILT.45 (G) In vivo ultrasound of the topical elastase/BAPN model at the point of maximal expansion, where yellow dashed lines represent the area of the AAA and the red dashed lines outline the ILT.
Composition of the ILT in humans and mouse models. (A) Representative picture of a 4.6-cm-diameter ILT (42.9 cm2) being removed from a 6-cm diameter aneurysm (79.4 cm2) during an open surgical procedure. (B) Macroscopic view of an ILT demonstrating the luminal, intermediate, and abluminal layers. (C) Ultrasound image of a human AAA with yellow and red dashed lines outlining the area of the aneurysm and thrombus, respectively. (D) A representative suprarenal AAA aorta from a low-density lipoprotein receptor–deficient (Ldlr−/−) mouse infused with AngII (1000 ng/kg per min) with an (E) in vivo ultrasound image captured at the maximum diameter on a Vevo 2100 (FUJIFILM VisualSonics Inc), where yellow dashed lines represent the area of the aneurysm and red dashed lines outline the extraluminal thrombus. (F) A representative infrarenal AAA aorta from a C57BL/6J mouse treated with topical elastase to induced aneurysm and administration of β3-aminopropionitrile fumarate salt (BAPN) ad libitum via water bottle to induce an ILT.45 (G) In vivo ultrasound of the topical elastase/BAPN model at the point of maximal expansion, where yellow dashed lines represent the area of the AAA and the red dashed lines outline the ILT.
The cellular and molecular mechanisms involved in the initiation, progression, and eventual rupture of AAA are not fully understood. Furthermore, even with advanced medical techniques and screening implementation programs to detect aneurysms, worldwide AAA mortality has not decreased.13 Understanding of these processes is needed to develop and implement pharmacological treatments for this condition. As the majority of AAAs are associated with the formation of a nonocclusive ILT within the aneurysm segment, antithrombotic therapies have emerged as potential pharmaceutical agents for the treatment of AAA progression. This perspective review describes the pathophysiology of AAA and ILT formation and the benefits and risks of antithrombotic therapy in the treatment of AAA.
Evolution of the AAA and the role of the coagulation cascade
Abdominal aortopathy involves 1 or more of the following processes: atherothrombosis (acquired), inflammation (acquired), or intrinsic collagen vascular disease (usually inherited). These etiologies manifest during several conditions, including acute and chronic infection, and connective tissue disorders such as Marfan syndrome, Ehler-Danlos syndrome, and Loeys-Dietz syndrome; however, the majority of AAAs are considered noninfectious inflammatory aneurysms with nonspecific causality.14,15 Although the initiating event in aneurysmal development is incompletely understood, elastin fiber degradation is 1 of the earliest events thought to occur.16 Degradation and exposure of elastin and underlying medial collagen result in VSMC activation, secretion of chemokines and cytokines, leukocyte recruitment and infiltration, and eventual endothelial activation and denudation, which likely is the nidus for ILT formation.11,14 Subsequent production, deposition, and activation of proteolytic enzymes, including plasmin-induced matrix metalloproteinases (MMPs), results in gelatinase, collagenase, and elastase activity accelerating elastin fragmentation and collagen degradation.17,18 Loss of elastin integrity, coupled with SMC apoptosis, is regarded as a key event in aneurysm formation, specifically increasing the abdominal diameter due to loss of structural integrity.19 As this occurs, the last line of defense is the type I and III–rich fibrillary collagen of the adventitia surrounding the aortic wall. As collagen turnover progresses (eg, increased catabolism vs production), the integrity of the adventitia weakens, which is speculated to be the ultimate cause of AAA rupture.16,20
AAAs are often associated with atherosclerotic damage of the aortic wall and are traditionally regarded as a consequence of atherosclerosis. However, several studies demonstrate aneurysms arise and progress in an alternative manner to athero-occlusive disease. Recently, the FAD (Fighting Aneurysmal Diseases) EU consortium classified AAA as an atherothrombotic disease, which unlike atherosclerosis, is characterized by local matrix-rich aortic medial destruction, presence of an ILT, and compensatory fibrosis in the adventitia.21 In particular, the biologically active ILT is a unique aspect of AAAs, whereby a thrombus is constantly exposed to flowing blood. Although the mechanism of vascular injury resulting in ILT formation remains unknown, it is speculated that injury to and subsequent loss of elastin (either by age or by inflammation) leads to elongation and tortuosity of the abdominal aorta causing asymmetric and turbulent flow profiles and abnormal wall stresses resulting in endothelial injury.22 As a result, vessel injury exposes matrix proteins, such as collagen and von Willebrand factor, to circulating platelets leading to activation via GPVI and GP1bα, respectively. Platelet activation results in release of platelet granules and autocrine activation of surrounding platelets via adenosine diphosphate and thromboxane A2 to propagate the platelet-rich plug. Together with simultaneous exposure of subendothelial tissue factor (TF) to blood and formation of the prothrombinase complex of factor Xa (FXa) and FVa, thrombin cleavage of fibrinogen to fibrin, and interaction of the primary hemostatic plug with FXIIIa crosslinked fibrin, a stable secondary platelet-fibrin thrombus forms. This beginning stage of the ILT eventually results in the formation of an organized thrombus that enlarges as a compensatory mechanism to offset the increasing diameter of the aorta to maintain pressure to conform to Laplace’s Law.
Nonocclusive trilayered ILT
Approximately 70% to 80% of AAA patients develop a nonocclusive ILT situated on the vessel wall, which permanently interfaces circulating blood (Figure 1A-C).21,23,24 The ILT is a documented determinant of AAA integrity and aneurysmal growth and may be biologically important.25,26 Early thrombus development constitutes a metabolically active structure and is characterized by the accumulation of activated platelets, small numbers of leukocytes (primarily neutrophils, known as polymorphonuclear cells), a thick fibrin mesh, and entrapped erythrocytes.23,27-29 Most thrombi contain a trilayered morphology, which originates from a continued temporal neutrophil apoptosis, erythrocyte lysis, and deposition of platelets and fibrin.30 This results in 3 regions of the ILT, referred to as the luminal (proximal to the blood), medial (intermediate region), and abluminal (proximal to the aneurysm wall) regions.23,29,30 The luminal layer exhibits a red hue (entrapment and hemolysis of erythrocytes) and is characterized by the presence of platelets, neutrophils, and a dense fibrin network, with secondary connections and crosslinks, up to a depth of 1 cm.29,30 The acellular medial layer contains loosely connected thin fibrin strands with no secondary connections and progressing matrix degradation.30 Finally, the abluminal layer contains poorly organized fibrin structures with completely degraded matrix and enhanced fibrinolytic activity. Small interconnected channels, termed “canaliculi,” are found throughout the ILT and become more prominent with increasing diameter toward the abluminal layer, allowing for unrestricted transportation of macromolecules (proteolytic enzymes, fluids, and cells) from the luminal to the abluminal layers.21,30,31 Moreover, as the aortic media are not vascularized and derive oxygen from diffusion across the lumen, hypoxia in the abluminal pole can exacerbate inflammation, fibrinolysis, and cell death of the adjacent aortic wall, resulting in progressive thinning of the wall and subsequent dilation, suggesting the thrombus is biologically active and contributes to the evolution of AAA pathogenesis.12,21,24
As a biologically active entity, the ILT acts as a source of secreted proteases, inflammatory cytokines, and fibrinolytic products within the layers of the thrombi, to the aneurysm wall via canaliculi, and into the bloodstream.28 Platelet accumulation and activation at the luminal edge of the thrombus result in secretion of MMPs, and a variety of thrombotic and inflammatory molecules, including platelet factor 4, interferon-γ, interleukin-1α, interleukin-β, and transforming growth factor-β.2,32 Deposition of the fibrin-containing macrophage antigen 1 (Mac-1/αMβ2/CD11b/CD18) ligand and exposure of platelet-derived P-selectin then result in the accumulation of neutrophils in the luminal layer of the ILT.23,28 Subsequent neutrophil degranulation and apoptosis result in the release of neutrophil extracellular traps, MMP-8, MMP-9, urokinase plasminogen activator (uPA), proteinase 3, cathepsins, myeloperoxidase (MPO), and elastase into the luminal layer.28,33 Circulating tissue plasminogen activator (tPA) and plasminogen bind to the free lysine residues exposed in luminal fibrin. Together, uPA and tPA result in the cleavage of plasminogen to plasmin resulting in the activation of released MMPs and transforming growth factor-β1, degradation of fibronectin, and cleavage of fibrin and resultant fibrinolysis in the luminal layer. Importantly, targeted disruption of MMP-9,34 MPO,35 uPA,36 and proteases33 attenuate the initiation of AAA in various animal models. Collectively, these molecules contribute to the inflammatory response and proteolytic injury of the arterial wall resulting in aortic thinning and expansion.28
The ILT contained in an AAA is unique due to a lack of temporal resolution/repair. Several studies have demonstrated a lack of reendothelialization, infiltration, and adherence of mesenchymal stem cells, and VSMC infiltration and survival into the layers of the ILT.21,23,28,30 These effects are likely due to the presence of neutrophils (capable of detaching endothelium from extracellular matrix),28 secretion of neutrophil elastase (degrades the key adherence protein fibronectin),28 and sundry proteases that prevent adhesion and growth of healthy cellular networks.30 Furthermore, MPO and erythrocyte lysis and release of free heme and iron produce a pro-oxidant environment, which augments damage to the aorta and thrombus in addition to reducing the survivability of infiltrating protective cells.21,35,37 Finally, association of exogenous weak pathogens, or commensal bacteria (eg, Porphyromonas gingivalis), is present in the aneurysm ILT, which may prevent wound healing via ongoing activation of platelets and repeated infiltration/activation of neutrophils via toll-like receptors.21 P gingivalis and Streptococcus mutans have been identified in aneurysm tissue, and the increased presence of P gingivalis is associated with augmented aneurysm and a neutrophil-rich ILT in a rat model of AAA.38,39 Together, the infiltration of inflammatory cells, release of inflammatory cytokines and proteolytic enzymes, and lack of wound healing result in elastin degradation and VSMC apoptosis and reduction in the aortic media leading to increased aortic weakness and dilation.
Evidence for the role of platelets in AAA
Platelets play an integral role in the formation, expansion, and proteolytic activity of the ILT.40,41 Surprisingly, very few publications have examined the role of platelets or platelet inhibitors on aneurysm pathophysiology in animal models and AAA patients. The 2 most widely used mouse models are the angiotensin II (AngII) infusion model and the elastase model of aneurysm (Figure 1D-G).1,34,42 As a caveat, animal models are limited by their lack of translational applicability to clinical pathology (reviewed in Sénémaud et al43 ). This is illustrated by a lack of spontaneous ILT formation in the majority of aneurysm models currently used. Although the AngII model does form a thrombus, it has minimal blood-thrombus interface occurring only at the point of medial dissection.44 Recent addition of BAPN (inhibitor of lysly oxidase) to the drinking water of the elastase aneurysm model does result in spontaneous ILT formation, similar to humans.45 Furthermore, although human aneurysms have an abundance of neutrophil infiltration (∼70% neutrophil contribution), most animal models are characterized by high monocyte/macrophage and lymphocyte infiltration (∼10% neutrophil contribution).46,47 However, although it is difficult to model a complex and heterogenous pathophysiology in animal models, these models are still useful to define mechanisms and pathways that may affect the human condition and provide complementary studies to test targeted therapeutics.
Abciximab, a glycoprotein IIb/IIIa inhibitor, attenuated thrombus area and prevented aortic enlargement in a rat xenograft model of aneurysm formation.29 This antiplatelet therapy decreased P-selectin expression, leukocyte binding to the luminal thrombus, and medial elastin degradation and increased the number of VSMCs adhering to the thrombus.29 Using this same model, administration of the P2Y12 receptor antagonist AZD6140 also decreased aortic diameter, thrombus development, leukocyte infiltration of the thrombus, elastic fiber degradation, and enhanced VSMC infiltration into the thrombus.46 In addition, this drug decreased CD41 and MMP-9 expression.46 Together, these results demonstrate a significant contribution of platelet activation to disease initiation, thrombus growth and renewal, and aneurysm progression in a rat model of AAA.
P-selectin deficiency attenuates aneurysm formation in the elastase-perfusion model.48 However, P-selectin is expressed on both endothelial cells and platelets; therefore, the contribution of endothelial compared with platelet-derived P-selectin is difficult to ascertain. The P2Y12 inhibitor clopidogrel reduced macrophage infiltration, reactive oxygen species formation, MMP-2, and aneurysm formation in the AngII aneurysm model.49 Our group investigated the effects of pharmacological inhibition of platelet activation on established AngII-induced aneurysms.2 The administration of ASA or clopidogrel reduced visible thrombi in the abdominal aorta, rupture-induced death, platelet and macrophage accumulation, MMP-2 and MMP-9 expression and activity, total and active uPA and tPA, and levels of platelet factor 4 and platelet-derived cytokines. Work from our laboratory demonstrates that platelet inhibition or depletion prior to aneurysm formation results in rupture-induced death in 3 different models of aneurysm (A.P.O., Marc Cambridge, Ziad Mallat, and Nigel Mackman, unpublished data, May 2012 to December 2017),50 suggesting platelet accumulation and activation may play a protective role in the early stages of aneurysm formation. Conversely, platelets may enhance inflammatory responses and secretion of proteolytic enzymes in late-stage aneurysms. This area requires further study regarding the timing of antiplatelet administration. Taken together, the literature suggests that platelets contribute to the progression of AAA.
AAA patients begin low-dose ASA at the time of diagnosis, because this may reduce all-cause mortality associated with cardiovascular disease (CVD).51,52 However, an in-depth analysis of ASA and other antiplatelet agents (P2Y12 inhibitors) are limited to retrospective analyses. Importantly, AAA patients have decreased platelet counts with increased levels of cleaved glycoprotein 1b, suggesting increased platelet activation and turnover vs control patients.41 Aneurysmal repair by EVAR resulted in reduced platelet counts and increased platelet activation immediately following surgery, which was correlated with endograft material, maximal diameter, and volume of the aneurysm.53 Last, morbidity and mortality are directly correlated with decreased platelet count upon admission to the hospital for emergency repair of ruptured AAAs or thrombocytopenia following repair.54 Interestingly, retrospective clinical trials and meta-analyses have reported no significant benefit of platelet inhibitors (ASA or P2Y12 inhibitors) on AAA progression or rupture in the clinic.2,55-58 Although low-dose ASA is not associated with increased risk of rupture in a large cohort of age- and sex-matched patients, ASA use is associated with a poor prognosis when given for the first 30 days postrepair.59 This result was not expected because antiplatelet therapy reduces 30-day and 5-year mortality after the incidence of major vascular surgeries (eg, carotid endarterectomy and open/endovascular AAA repair).60 Despite these findings, our group evaluated the progression and rupture of AAA in patients with or without ASA or P2Y12 antagonists and demonstrated a significant decrease in mortality.2 Furthermore, low-dose ASA use reduced aneurysm expansion rate and risk of future surgical repair in AAAs measuring 4.0 to 4.9 cm.61 However, no difference was found in patients with aneurysms <4 cm, and the total population of patients was relatively small. Finally, low-dose ASA is associated with decreased thrombus volume (determined by computed tomographic scan) in patients undergoing elective repair. However, these effects were not demonstrated with statins, angiotensin-converting enzyme inhibitors, and clopidogrel usage. This suggests that the effect may be due to the anti-inflammatory effect of ASA and not the antiplatelet properties. Together, the data from animal studies and clinical observations demonstrate that platelets play an important role in aneurysm formation, progression, and rupture. Although most data suggest antiplatelet drugs may have no effect, the use of antiplatelet drugs to deter other cardiovascular morbidities and mortalities is still warranted. As most studies used ASA, further studies need to be conducted with newer classes of antiplatelet therapies.
Evidence for the role of coagulation in AAA
Coagulation and fibrin deposition are crucial to the formation and expansion of the ILT. However, there are few studies examining the role of the coagulation cascade and alternatively focus on the fibrinolytic pathway and fibrin degradation products. Interventional administration of fondaparinux (synthetic, a selective FXa inhibitor) or enoxaparin (low-molecular-weight heparin that also inhibits FXa), 14 days post-AngII infusion, reduced endpoint aneurysm diameter compared with placebo control.62 Fondaparinux reduced PAR-2 and MMP-2 gene expression, elastic fiber fragmentation, monocyte/macrophage infiltration, and aortic diameter compared with controls. Enoxaparin significantly reduced aortic diameter compared with controls. Interestingly, the administration of dabigatran, a direct thrombin inhibitor, did not significantly influence aneurysm growth. However, comparison of different anticoagulants is difficult because they may have different levels of inhibition in the model. Unpublished data from our laboratory demonstrates that administration of dabigatran or rivaroxaban (FXa inhibitor) prior to aneurysm formation results in increased rupture-induced death in an AngII model (A.P.O., James Luyendyk, and Nigel Mackman, unpublished data, July 2014 to May 2017). Similar to our antiplatelet data, this suggests that impairment of hemostasis prior to aneurysm formation is detrimental to stability perhaps due to loss of vascular integrity without the ability to develop a primary and secondary hemostatic plug.
Although the majority of coagulation-based mouse studies in AAA have focused on the role of the fibrinolytic pathway, some results are contradictory. Deletion of the enzyme thrombin-activatable fibrinolysis inhibitor (also called procarboxypeptide B) augments aneurysm diameter, rupture-induced death, and plasmin generation in the elastase model.63 Because thrombin-activatable fibrinolysis inhibitor prevents plasmin cleavage of fibrin and results in clot stability, the preceding paper suggests clot stability is imperative in the elastase model. Furthermore, uPA and overexpression of plasminogen activator inhibitor 1, respectively, attenuate the formation of AAA formation in the AngII model, suggesting increased fibrinolysis and plasmin formation increased aneurysm size and rupture.36,64 However, uPA and uPA receptor deficiency has no effect on AngII-induced AAA, and that uPA deficiency augments rupture-induced death in a hematopoietic manner.65 Although thrombolysis clearly has a role in aneurysm progression, the mechanism is still being defined.
A role for the coagulation cascade in human AAAs is observed via elevated TF and markers of thrombin formation, fibrin formation, and fibrin turnover in AAA patients. Patients undergoing emergency AAA repair have increased plasma TF activity vs AAA patients undergoing elective repair utilizing the ACTICHROME TF assay.66 However, it must be noted that the ACTICHROME TF assay has several limitations, because FXa activity can be measured without exogenous FVIIa or TF; therefore, these results should be interpreted with caution.67 TF is also present in all 3 layers of the ILT in human AAA.29 A recent study suggests human AAAs have an altered clot structure independent of plasma fibrinogen, thrombin generation, and circulating TF; however, the number of patients analyzed in this study was small.68 Thrombin (as measured by thrombin-antithrombin) is increased in patients with AAAs,68-73 and levels correlate with the maximum diameter of the aorta in AAA patients.70,72,73 A recent meta-analysis demonstrated elevation of multiple hemostatic indicators, including D-dimer and thrombin-antithrombin in AAA patients.71 A prospective observational study demonstrated a strong positive correlation between increasing D-dimer and AAA progression.74 Levels of naturally occurring anticoagulants, such as soluble thrombomodulin (TM), have a significant role in the inflammation and tissue damage associated with aneurysm formation.75 Delivering recombinant TM domains confers anti-inflammatory protection and reduction of aneurysm formation.76 Furthermore, TM exerts proinflammatory effects on macrophages utilizing cell-specific TM deletions in the calcium chloride model.77 Taken together, the results of animal studies and consistent elevation of established thrombotic markers in AAA patients indicate an important contribution of coagulation to the progression of AAA.
Interpreting the consequences of oral anticoagulant use in patients with AAA is challenging. High-quality clinical trials showed a mortality benefit in using direct thrombin inhibitors (ROCKET AF trial) and an Xa inhibitor (COMPASS trial), although the exact number of patients in these trials with concomitant PVD and AAA is unclear.78,79 Conversely, EVAR-repaired AAAs with concomitant administration of vitamin K antagonists result in type II endoleaks. This involves aortic branch bleeding into the aneurysmal sac below the aortic wall where the stent is opposed, implying the coagulation cascade may be protective in the early stages of aortic wall integrity following EVAR.80 A larger meta-analysis demonstrated that long-term anticoagulation in EVAR patients was associated with persistent type II endoleaks.81 In aortopathies, thrombosis and hemorrhage may manifest in the lumen as well as within the intraluminal region of the vasculature. Development of an intramural hematoma within the wall of the aorta constitutes a true acute aortic syndrome with treatment similar to ruptured AAAs with published data showing anticoagulants are detrimental and may precipitate IHM in this clinical scenario.82,83 Conversely, other studies have shown enzymes such as MMP-9 are inhibited by FXa antagonists, low-molecular-weight heparin, and vitamin K antagonist use and may promote sac integrity surrounding a stent in a type II endoleak.80,84
Nonocclusive mural thrombus and aortic wall stability
The volume of the AAA ILT strongly correlates with aneurysmal size and growth.62,85,86 An underlying assumption exists that a thrombus smaller in size would be beneficial to long-term patient outcomes, because it would correlate with a smaller aneurysm and lesser risk of rupture.87 However, evidence suggests that AAA rupture is a result of mechanical stress on the aortic wall.88 The thrombi located within an aneurysmal sac can likely alleviate stress on the arterial wall and protect against rupture.89
Researchers have attempted to use imaging and physical modeling to calculate the true relationship of thrombus size and risk of rupture. Through imaging and mathematical models, researchers can calculate the mechanical stress on the aortic wall, given placement and size of the thrombus.89 Although imaging improves assessment of individual risk of rupture, it does not definitively define risk based on thrombus size alone. Studies have suggested that an increase in thrombus diameter may decrease stress on the aortic wall and thus decrease rupture, whereas others have failed to find either a proportional increase or a decrease in mechanical stress with an increase in thrombus size.90,91 The thrombus may act as a mechanical cushion, reducing the risk for fatal ruptures, although asymmetrical thrombi may increase the total stress on the aorta wall.89 Of note, this effect is particularly dependent on the placement of the thrombus within the aorta. Moreover, the aortic wall surrounding the thrombus may be weakened by aortic wall hypoxia at the site of thrombus attachment.92 This is supported by autopsy findings of patients with ruptured aneurysms wherein the majority of aneurysms ruptured at the ILT.93 As a point of comparison, there is a recurring phenomenon in vascular medicine regarding aortic dissection as a temporary containment of the dissection flap by thrombotic material, which protects the entry point for blood into a false lumen preventing subsequent propagation of the dissection. Thrombus removal, without a plan for repair, can have catastrophic consequences with fulminant aortic rupture, generally a situation resulting in patient mortality. As such, there are compelling reasons not to therapeutically reduce thrombus size without first evaluating aortic wall stability.
The ILT in AAA is self-perpetuating. Although the coagulation cascade is natively proficient at amplifying a clot (necessary for hemostasis), it also propagates an inflammatory response and proteolytic injury at the arterial wall. As expected, a larger thrombus is associated with a higher overall AAA growth rate.91 However, in thrombotic events, this results in an ever-growing, problematic clot and could lead to occlusion (rare) or AAA rupture due to mere size. Ultimately, the benefit of the nonocclusive ILT is dependent on placement in the aorta and is best evaluated on a patient-specific basis taking into account mechanical stress and occlusion in each situation.
Antithrombotic therapy: beneficial or detrimental?
AAA is a complex disorder lacking pharmacological therapies and noninvasive treatment options. Although EVAR has greatly improved postsurgical outcomes, morbidity and mortality remain high in these patients.94 With new knowledge regarding the role of platelet activation and the coagulation cascade in AAA pathogenesis, antithrombotic therapy, including antiplatelet drugs, anticoagulants, and thrombolytics, has been considered. As previously stated, antiplatelet drugs have significantly reduced AAA burden in animal models but have failed to consistently produce significant results clinically. Similarly, anticoagulant drugs have shown potential in preventing AAA progression in animal models, but clinical analysis is limited, and patients on oral anticoagulant therapy for other comorbid conditions have not shown advantage over other patients with AAA not using oral anticoagulants.57 Anticoagulant drugs also present great risk of bleeding in the event of rupture, discouraging clinical use.95,96 Currently, neither antiplatelet nor anticoagulant drugs are clinically used aside from ASA therapy recommended as secondary prevention for the risk of CVD.2,6,9
Given the preponderance of data, the decision to initiate antithrombotic therapy in the setting of AAA is a patient-specific decision that should consider aortic wall injury, stability, and growth rate. Although the thrombus seems to contribute to the size, growth, and proteolytic injury of the arterial wall, it may decrease mechanical stress on the aortic wall and maintain AAA stability. Although antithrombotic therapy could reduce proteolytic injury, it could concurrently reduce mechanical stability of the aneurysm. A balance must be struck between the detrimental and protective contributions of the thrombus to aneurysm progression.
Although one-size-fits-all guidelines provide clinicians with clear recommendations for patients, this complex disease may require precision medicine approaches. Using advanced imaging and physical modeling techniques, clinicians are able to assess thrombus size and mechanical stress on the aortic wall. In addition, nuclear medicine imaging techniques can be used to assess the makeup and biological effects of the ILT.97 As D-dimer is currently used as a diagnostic test for acute aortic syndromes, including aortic dissection, measures of thrombotic and inflammatory markers may provide additional evidence of thrombus status in AAA patients.98 The risk of rupture must be carefully considered after measurement of these variables. If a patient’s condition indicates that the thrombus is unstable, anticoagulants may be considered to reduce thrombus size, proteolytic injury of the aortic wall, and aneurysm growth. These questions need to be assessed with hypothesis-driven experiments and clinical outcomes to determine if thrombus composition can be used as a predictive indicator of aneurysm stability or eminent wall failure and rupture.
Conclusion
AAA is a complex thrombotic and inflammatory disorder. Platelet activation and coagulation play crucial roles in the development of the nonocclusive ILT, progression of aneurysm development, and eventual rupture. Surgical intervention is the only treatment option, and there is a great need for pharmacological prevention for AAA patients.
Because antiplatelet therapy is standard for most CVDs and thrombotic disorders, ASA therapy has been used in AAA with retrospective analyses showing no improvement in patient outcomes.57 Nevertheless, animal models have demonstrated improvement with antiplatelet drugs such as abciximab and P2Y12 inhibitors.2,29 Abciximab is only available for parenteral administration; therefore, outpatient management is not practical. This demonstrates a potential for further investigation into the development of orally available antiplatelet therapies in addition to ASA, likely necessitating identification of additional platelet targets to modulate aneurysm growth and rupture.
Although some animal studies have demonstrated success with anticoagulant therapies, anticoagulants are not currently used to treat AAA.62 Reservations persist, because AAA usually terminates in a rupture and ultimately a hemostatic crisis if surgical intervention is not performed. Because anticoagulant therapies are notoriously difficult to reverse in the event of hemorrhage, anticoagulants have not been traditionally considered in cases of AAA. However, some researchers have proposed use of reversible anticoagulants by using RNA aptamers.99 Anticoagulant therapy may be worth investigating; however, reversible anticoagulants are in early development and currently only available in administrations best considered in inpatient settings.
Considering the role of the thrombus in aneurysmal diameter and progression and the ILT in the stabilization of AAA, anticoagulant therapy must be carefully considered. Proposed mathematical models could suggest patient-specific analysis of individual AAAs.89 If these aneurysms are not stabilized by the thrombus, then anticoagulant therapy could be beneficial. Anticoagulant therapy may also be warranted in patients with occlusive aneurysm. As such, it is necessary to build criteria that better reflect the individual status of a patient’s ILT. In addition, clinical trials investigating the benefit of anticoagulation or wall stress analysis should be pursued. With the discovery of rapidly reversible anticoagulants, treatment of thrombus formation in a patient-specific manner could result in significant reductions in morbidity and mortality for AAA patients.
Although the study of anticoagulant therapy is limited and such therapy has not been used clinically to treat AAA, advanced imaging and physical modeling techniques and assessment of thrombus composition and biological affects make it possible to better assess thrombus and aneurysm status, providing data to individualize patient care and recommend anticoagulant therapy to specific AAA patients (Figure 2). With a great need for pharmacological treatment of this disease, this patient-specific therapy holds great potential.
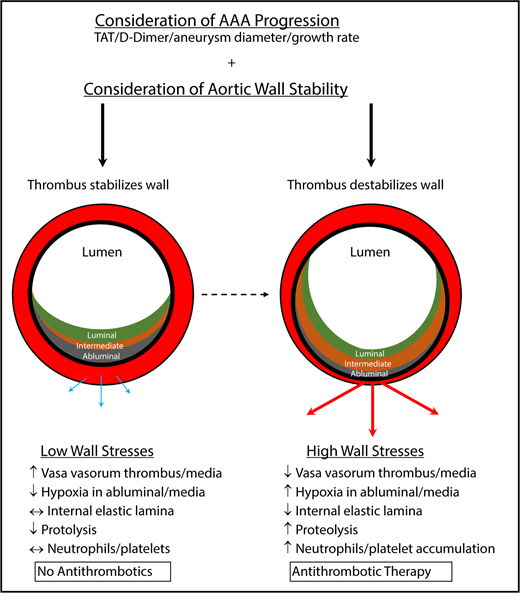
Determinations for antithrombotic treatment in patients with AAAs. Antithrombotic therapy, including antiplatelet drugs and anticoagulants, should likely be determined on a patient-to-patient basis. To assess whether patients should receive antithrombotic therapy, AAA progression and aortic wall stability should be assessed. With nominal aortic diameter growth, progression, and low wall stresses, antithrombotics should not be considered because the ILT may protect the wall from further harm. However, a destabilized thrombus with rapidly expanding aortic diameter, progression, and increased wall stress may benefit from antithrombotic therapy to prevent additional degradation of the vasculature, further weakening the aorta and resulting in rupture. Blue/thin arrows represent low outward force, whereas red/thick arrows represent high outward force. TAT, thrombin-antithrombin.
Determinations for antithrombotic treatment in patients with AAAs. Antithrombotic therapy, including antiplatelet drugs and anticoagulants, should likely be determined on a patient-to-patient basis. To assess whether patients should receive antithrombotic therapy, AAA progression and aortic wall stability should be assessed. With nominal aortic diameter growth, progression, and low wall stresses, antithrombotics should not be considered because the ILT may protect the wall from further harm. However, a destabilized thrombus with rapidly expanding aortic diameter, progression, and increased wall stress may benefit from antithrombotic therapy to prevent additional degradation of the vasculature, further weakening the aorta and resulting in rupture. Blue/thin arrows represent low outward force, whereas red/thick arrows represent high outward force. TAT, thrombin-antithrombin.
Acknowledgments
The authors thank Doran Mix (University of Rochester Medical Center) for his hard work and photographs of human AAAs. They also thank Nigel Mackman (University of North Carolina at Chapel Hill), Kelsey Conrad (University of Cincinnati), Keith Saum (University of Cincinnati), Sarah Anthony (University of Cincinnati), and Michael Tranter (University of Cincinnati) for thoroughly proofreading this article and providing useful comments.
This work is supported by National Institutes of Health, National Heart, Lung, and Blood Institute grants 5R00-HL116786-06 and 1R01-HL141401-01 (A.P.O.), 5T32HL066988-1, 2K08HL128856, and HL12020 (S.J.C.), and F31 HL143987-01 (H.M.R.); and the Sable Heart Fund (Independent Order of Oddfellows) and a University of Rochester Department of Medicine Pilot grant (S.J.C.).
Authorship
Contribution: S.J.C., H.M.R., and A.P.O. contributed to the writing of the manuscript; A.P.O. and S.C. made Figure 1; H.M.R. made Figure 2; and all authors reviewed and corrected both figures.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: A. Phillip Owens III, University of Cincinnati, 231 Albert Sabin Way, ML: 0542, Cincinnati, OH 45267-0542; e-mail: phillip.owens@uc.edu.
