

Key Points
Sos1 mediates oncogenic Kras-induced WT Nras and Hras hyperactivation.
Sos1−/− attenuates KrasG12D-induced MPN and prolongs the survival of KrasG12D mice.

Abstract
We and others have previously shown that KrasG12D is a much more potent oncogene than oncogenic Nras in hematological malignancies. We attributed the strong leukemogenic activity of KrasG12D at least partially to its unique capability to hyperactivate wild-type (WT) Nras and Hras. Here, we report that Sos1, a guanine nucleotide exchange factor, is required to mediate this process. Sos1 is overexpressed in KrasG12D/+ cells, but not in NrasQ61R/+ and NrasG12D/+ cells. KrasG12D proteins form a complex with Sos1 in vivo. Sos1 deficiency attenuates hyperactivation of WT Nras, Hras, and the downstream ERK signaling in KrasG12D/+ cells. Thus, Sos1 deletion ameliorates oncogenic Kras-induced myeloproliferative neoplasm (MPN) phenotypes and prolongs the survival of KrasG12D/+ mice. In contrast, Sos1 is dispensable for hyperactivated granulocyte-macrophage colony-stimulating factor signaling in NrasQ61R/+ cells, and Sos1−/− does not affect MPN phenotypes in NrasQ61R/+ mice. Moreover, the survival of KrasG12D/+; Sos1−/− recipients is comparable to that of KrasG12D/+ recipients treated with combined MEK and JAK inhibitors. Our study suggests that targeting Sos1-oncogenic Kras interaction may improve the survival of cancer patients with KRAS mutations.
Introduction
Mammalian Ras genes (Hras, Nras, and Kras) encode small GTPases that cycle between active GTP-bound form and inactive GDP-bound form.1 The conversion from the GDP-bound form to the GTP-bound form is facilitated by guanine nucleotide exchange factors, such as Sos1. Canonical, cancer-associated RAS mutations at codons G12, G13, or Q61 severely compromise the hydrolysis of GTP to GDP, leading to the accumulation of Ras-GTP and hyperactivation of Ras downstream signaling (eg, RAF/MEK/ERK pathway). Mutations in RAS/ERK pathway genes (including KRAS, NRAS, CBL, NF1, and PTPN11) are well known to drive the pathogenesis of juvenile myelomonocytic leukemia.2,3 In addition, a recent genomic study identified prevalent RAS/ERK pathway mutations in pediatric myelodysplastic syndrome,4 highlighting the emerging role of RAS pathway mutations in pediatric myeloid malignancies.
Despite their similar expression patterns and high protein sequence similarities, we found that KrasG12D is a much more potent oncogene than oncogenic Nras; expression of KrasG12D, through either pI-pC induction or leaky expression of Mx1-Cre, drives an acute myeloproliferative neoplasm (MPN) and leads to much earlier lethality than oncogenic Nras in mice (reviewed in Chang et al5 ). Consistently, juvenile myelomonocytic leukemia with heterozygous somatic KRAS mutations are particularly aggressive and generally require an urgent bone marrow transplantation therapy.6 We attributed the strong leukemogenic activity of KrasG12D at least partially to its unique capability to hyperactivate wild-type (WT) Nras and Hras.7 Previous crystallographic analyses of Ras/Sos complex showed that Ras-GTP binds to the catalytic domain of Sos at a site distal to the active site of Sos, and stabilizes this active site allosterically.8 Thus, it was hypothesized that the interaction between Ras-GTP and Sos1 may accelerate Sos-stimulated GTP upload on empty Ras. Subsequently, Bar-Sagi’s group reported that in cancer cell lines, overexpression of oncogenic Kras cross-activates WT Nras and Hras, and that this process is mediated by Sos1.9 Here, we officially tested this hypothesis in KrasG12D and NrasQ61R mice in vivo.
Methods
Mice
All mouse lines were maintained in a pure C57BL/6 genetic background (>N10). All animal experiments were conducted in accordance with the Guide for the Care and Use of Laboratory Animals and approved by an Animal Care and Use Committee at the University of Wisconsin-Madison. The program is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care.
Results and discussion
Sos1 deletion abolishes oncogenic Kras-induced hyperactivation of WT Nras and Hras
To test our hypothesis that Sos1 mediates oncogenic Kras-induced WT Nras and Hras hyperactivation, but is dispensable for oncogenic Nras, we first confirmed that Sos1 was overexpressed in KrasG12D/+ bone marrow cells (P < .01; Figure 1A), but not in NrasQ61R/+ and NrasG12D/+ cells (Figure 1B; supplemental Figure 1A, available on the Blood Web site). Immunoprecipitation of KrasG12D protein using anti-Ras G12D antibody pulled down Sos1 protein (Figure 1C), whereas immunoprecipitation of NrasG12D protein did not (supplemental Figure 1B). These results indicate that KrasG12D, but not NrasG12D, forms a complex with Sos1 in vivo. We then generated compound KrasLSL G12D/+;Sos1fl/fl;Vav-Cre mice10 (referred to as Kras;Sos1−/− mice thereafter) with Vav-Cre-mediated KrasG12D expression and Sos1 deletion. Unlike the Vav-iCre that drives oncogenic Kras expression in both fetal liver hematopoietic and endothelial cells, and thus leads to an embryonic lethality,11 the Vav-Cre we used in our study12,13 drove oncogenic Kras expression more strictly in the hematopoietic system after embryonic day 11.5, and thus approximately two-thirds of KrasLSL G12D/+;Vav-Cre (Kras) mice survived until adulthood (supplemental Table 1). As a consequence, approximately two-thirds of Kras;Sos1−/− mice also reached adulthood (supplemental Table 2).
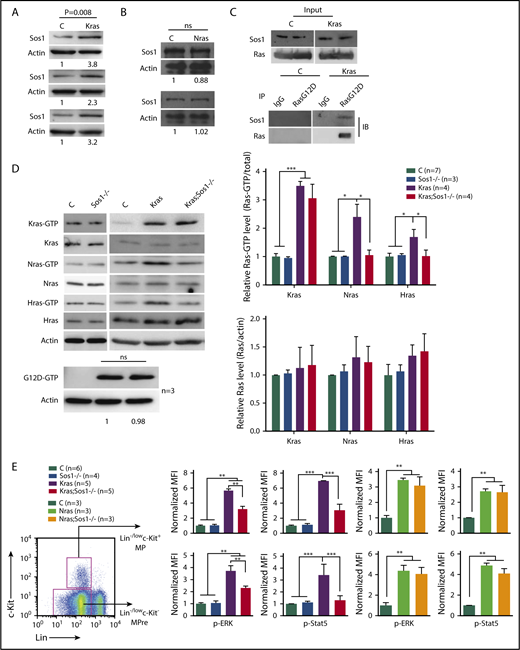
Sos1 loss abolishes activation of WT Nras and Hras and decreases GM-CSF signaling in KrasG12D/+cells. Control (C), Sos1fl/fl;Vav-Cre (Sos1−/−), KrasLSL G12D/+;Vav-Cre (Kras), KrasLSL G12D/+;Sos1fl/fl;Vav-Cre (Kras; Sos1−/−), NrasLSL Q61R/+;Vav-Cre (Nras), and NrasLSL Q61R/+;Sos1fl/fl;Vav-Cre (Nras; Sos1−/−) mice were sacrificed at age 6 weeks. (A-B) Sos1 expression levels in bone marrow (BM) cells of control, Kras (A), and Nras (B) mice were quantified against the levels of β-actin, using ImageStudioLite software. The ratios in control cells are arbitrarily set at 1. ns, not significant. (C) Whole-cell lysates were extracted from BM cells and immunoprecipitated with anti-Ras G12D antibody or preimmune immunoglobulin G (IgG). The resulting precipitates were immunoblotted with anti-Sos1 or anti-Ras antibody. (D) Whole-cell lysates were extracted from BM cells and analyzed for expression levels of different Ras isoforms, which were quantified against the levels of β-actin, using ImageStudioLite software. Ras-GTP was affinity purified from whole-cell lysates, using a GST fusion with the Ras binding domain of Raf (Raf RBD) immobilized on agarose beads. The levels of Ras-GTP bound forms were quantified against the levels of their corresponding Ras isoforms. The ratios of Ras-GTP/Ras in control cells are arbitrarily set at 1. Oncogenic Kras was detected using an antibody specifically against Ras G12D protein and quantified against the levels of β-actin. (E) BM cells were serum- and cytokine-starved for 2 hours at 37°C. Cells were then stimulated with 2 ng/mL mGM-CSF for 10 minutes at 37°C. Levels of p-ERK1/2 and p-STAT5 were measured using phosphoflow cytometry. Myeloid progenitors (MP) are enriched in Lin−/low c-Kit+ cells and myeloid precursors (MPre) are enriched in Lin−/low c-Kit− cells. Data are presented as mean + standard deviation. *P < .05; **P < .01; ***P < .001.
Sos1 loss abolishes activation of WT Nras and Hras and decreases GM-CSF signaling in KrasG12D/+cells. Control (C), Sos1fl/fl;Vav-Cre (Sos1−/−), KrasLSL G12D/+;Vav-Cre (Kras), KrasLSL G12D/+;Sos1fl/fl;Vav-Cre (Kras; Sos1−/−), NrasLSL Q61R/+;Vav-Cre (Nras), and NrasLSL Q61R/+;Sos1fl/fl;Vav-Cre (Nras; Sos1−/−) mice were sacrificed at age 6 weeks. (A-B) Sos1 expression levels in bone marrow (BM) cells of control, Kras (A), and Nras (B) mice were quantified against the levels of β-actin, using ImageStudioLite software. The ratios in control cells are arbitrarily set at 1. ns, not significant. (C) Whole-cell lysates were extracted from BM cells and immunoprecipitated with anti-Ras G12D antibody or preimmune immunoglobulin G (IgG). The resulting precipitates were immunoblotted with anti-Sos1 or anti-Ras antibody. (D) Whole-cell lysates were extracted from BM cells and analyzed for expression levels of different Ras isoforms, which were quantified against the levels of β-actin, using ImageStudioLite software. Ras-GTP was affinity purified from whole-cell lysates, using a GST fusion with the Ras binding domain of Raf (Raf RBD) immobilized on agarose beads. The levels of Ras-GTP bound forms were quantified against the levels of their corresponding Ras isoforms. The ratios of Ras-GTP/Ras in control cells are arbitrarily set at 1. Oncogenic Kras was detected using an antibody specifically against Ras G12D protein and quantified against the levels of β-actin. (E) BM cells were serum- and cytokine-starved for 2 hours at 37°C. Cells were then stimulated with 2 ng/mL mGM-CSF for 10 minutes at 37°C. Levels of p-ERK1/2 and p-STAT5 were measured using phosphoflow cytometry. Myeloid progenitors (MP) are enriched in Lin−/low c-Kit+ cells and myeloid precursors (MPre) are enriched in Lin−/low c-Kit− cells. Data are presented as mean + standard deviation. *P < .05; **P < .01; ***P < .001.
To determine whether Sos1 deletion affects WT Ras activation in the absence or presence of oncogenic Kras, we analyzed levels of Ras-GTP bound forms in bone marrow cells isolated from 6-week-old control (Vav-Cre), Sos1−/−, Kras, and Kras;Sos1−/− mice. In the absence of oncogenic Kras, Sos1 deficiency did not affect expression of different Ras isoforms, nor did it significantly affect Ras-GTP levels (Figure 1D). Granulocyte-macrophage colony-stimulating factor (GM-CSF)-stimulated ERK and STAT5 activation in Sos1−/− cells were comparable to those in WT cells (Figure 1E). Our result is consistent with a previous report that Sos1 is dispensable for mouse survival, and its function is likely to be compensated by other Ras guanine nucleotide exchange factors.14 In contrast, in Kras cells, Sos1 deficiency abolished hyperactivation of WT Nras and Hras without a significant effect on total Kras-GTP or KrasG12D-GTP level (Figure 1D). Associated with downregulation of WT Nras and Hras activation, GM-CSF-stimulated ERK and STAT5 activation were also greatly reduced in Kras;Sos1−/− cells compared with Kras cells (Figure 1E). Given the universal expression of Kras, Nras, Hras, and Sos1 in all populations of myeloid progenitors (https://gexc.riken.jp and our RNA-Seq data in Kong et al15 ), we believe that the effect of Sos1 is applicable to all these cells.
We also generated NrasLSL Q61R/+;Vav-Cre (Nras) and NrasLSL Q61R/+;Sos1fl/fl;Vav-Cre (Nras;Sos1−/−) mice. Consistent with our previous observation that oncogenic Nras does not lead to hyperactivation of WT Kras and Hras,7,16 GM-CSF-stimulated ERK and STAT5 activation in Nras;Sos1−/− cells was comparable to that in Nras cells (Figure 1E). Together, our data suggest that Sos1 mediates oncogenic Kras-induced WT Nras and Hras hyperactivation in vivo, whereas Sos1 function is dispensable for hyperactivated GM-CSF signaling in NrasQ61R/+ cells. We believe that the differential dependence on Sos1 is mainly a result of the divergent C-terminal highly variable regions of Nras and Kras proteins, which are known to regulate their distinct localization on the plasma membrane.17
Sos1 deficiency attenuates oncogenic Kras-induced MPN phenotypes and prolongs the survival of Kras mice
Given our cellular and signaling studies described here, we investigated whether Sos1 deletion attenuates oncogenic Kras-induced and/or oncogenic Nras-induced MPN phenotypes, which are characterized by increased white blood cell count and marked splenomegaly. Although the MPN phenotypes in Nras; Sos1−/− mice were comparable to those in Nras mice (supplemental Figure 2), these phenotypes were partially rescued in Kras;Sos1−/− mice (Figure 2A-B). Hematoxylin and eosin staining of Kras spleens showed great expansion of the red pulp with extensive extramedullary hematopoiesis consisting of erythroid, megakaryocytic, and myeloid precursor cells (Figure 2C). In contrast, Kras;Sos1−/− spleens demonstrated relative preservation of splenic architecture with only a mild increase in extramedullary hematopoiesis. As we expected, Kras;Sos1−/− mice survived significantly longer than Kras mice (Figure 2D). Both Kras and Kras;Sos1−/− mice developed a fully penetrant MPN, and approximately 50% of Kras mice and 40% of Kras;Sos1−/− mice simultaneously developed T-cell acute lymphoblastic leukemia/lymphoma (Figure 2E). We genotyped bone morrow cells from moribund Kras;Sos1−/− mice and showed that Sos1 remained deleted in these cells (Figure 2F). The absence of Sos1 protein was confirmed in Sos1−/− and Kras;Sos1−/− cells, using western blot analysis (Figure 2G).

Loss of Sos1 alleviates MPN phenotypes and prolongs the survival of oncogenic Kras mice in the Vav-Cre system. (A-C) Control (C), Sos1fl/fl;Vav-Cre (Sos1−/−), KrasLSL G12D/+;Vav-Cre (Kras), and KrasLSL G12D/+;Sos1fl/fl;Vav-Cre (Kras; Sos1−/−) mice were sacrificed at age 6 weeks. (A) Numbers of WBC (white blood cells) and PLT (platelets) were shown. (B) Quantification of spleen weight. (C) Representative histologic hematoxylin and eosin sections of spleen from Control, Sos1−/−, Kras, and Kras;Sos1−/− mice. (D-E) Kras and Kras;Sos1−/− mice were monitored closely until a moribund stage for terminal analysis. (D) Kaplan-Meier survival curves of different groups of mice were plotted against days after birth. P values were determined using the Log-rank test. (E) Quantification of disease incidence. χ-square analysis was performed. ns, not significant. (F-G) Moribund Kras and Kras;Sos1−/− mice and age-matched control (C) and Sos1−/− mice were sacrificed for analysis. (F) Genomic DNAs were extracted from tail and BM cells and genotyped for Sos1 alleles. (G) Western blot analysis of Sos1 expression in BM cells. The results are presented as mean ± standard deviation. *P < .05; **P < .01; ***P < .001.
Loss of Sos1 alleviates MPN phenotypes and prolongs the survival of oncogenic Kras mice in the Vav-Cre system. (A-C) Control (C), Sos1fl/fl;Vav-Cre (Sos1−/−), KrasLSL G12D/+;Vav-Cre (Kras), and KrasLSL G12D/+;Sos1fl/fl;Vav-Cre (Kras; Sos1−/−) mice were sacrificed at age 6 weeks. (A) Numbers of WBC (white blood cells) and PLT (platelets) were shown. (B) Quantification of spleen weight. (C) Representative histologic hematoxylin and eosin sections of spleen from Control, Sos1−/−, Kras, and Kras;Sos1−/− mice. (D-E) Kras and Kras;Sos1−/− mice were monitored closely until a moribund stage for terminal analysis. (D) Kaplan-Meier survival curves of different groups of mice were plotted against days after birth. P values were determined using the Log-rank test. (E) Quantification of disease incidence. χ-square analysis was performed. ns, not significant. (F-G) Moribund Kras and Kras;Sos1−/− mice and age-matched control (C) and Sos1−/− mice were sacrificed for analysis. (F) Genomic DNAs were extracted from tail and BM cells and genotyped for Sos1 alleles. (G) Western blot analysis of Sos1 expression in BM cells. The results are presented as mean ± standard deviation. *P < .05; **P < .01; ***P < .001.
We further validated our mouse results using a different Cre, Mx1-Cre, to mediate oncogenic Kras expression and Sos1 deletion. Consistent with our data from the Vav-Cre system, we also observed reduced splenomegaly (supplemental Figure 3A) in 6-week-old mice and prolonged survival by Sos1 deletion (supplemental Figure 3B) in the Mx1-Cre system. Our results indicate that Sos1 deficiency attenuates KrasG12D-induced MPN phenotypes and prolongs the survival of Kras mice.
Sos1 deletion and combined inhibition of MEK and JAK prolong the survival of Kras recipients
At this time, small compounds targeting oncogenic Kras-Sos1 interaction in vivo are not available. Therefore, we compared the effect of Sos1 deficiency with that of combined MEK and JAK inhibition on oncogenic Kras-induced leukemogenesis. We used AZD6244, a potent MEK inhibitor,18 and CYT387, a JAK1/2 inhibitor that strongly interrupts an autocrine cytokine circuit in a Kras-driven lung cancer model.19 Combined AZD6244 and CYT387 treatment effectively inhibited the growth of mouse Kras MPN cells (supplemental Figure 4A) and human juvenile myelomonocytic leukemia cells (supplemental Figure 4B). For the in vivo study, we established 2 recipient cohorts: one transplanted with Kras splenocytes and the other with Kras;Sos1−/− splenocytes. Both cohorts were analyzed at 4 weeks after transplantation. Despite their similar donor-derived contribution in the peripheral blood of recipients (supplemental Figure 4C), the average percentage of donor-derived monocytes in Kras;Sos1−/− recipients was lower than that in Kras recipients (supplemental Figure 4D), indicating a milder disease progression. We further divided each cohort into 2 groups and treated them with vehicle or combined AZD6244 and CYT387 until a moribund stage. Both groups showed comparable donor contribution and donor-derived monocytes within each cohort (supplemental Figure 4C-D). Compared with vehicle treatment, combined treatment of AZD6244 and CYT387 prolonged the survival of Kras recipients, which was comparable to the survival of Kras;Sos1−/− recipients treated with vehicle (supplemental Figure 4E). Combined treatment further prolonged the survival of Kras;Sos1−/− recipients without significantly changing the disease incidence (supplemental Figure 4F). These data suggest that the interaction between Sos1 and oncogenic Kras is a potential therapeutic target, and disruption of this interaction could be combined with other therapies for treating oncogenic Kras-driven cancers.
In summary, we showed the unique dependence on Sos1 in oncogenic Kras-induced leukemogenesis. In Kras mice, oncogenic Kras and hyperactivated WT Nras and Hras via Sos1 cooperate to drive MPN. In contrast, in Kras;Sos1−/− mice, WT Nras and Hras hyperactivation is attenuated and MPN is promoted by oncogenic Kras signaling alone (supplemental Figure 4G). Therefore, Sos1 deletion ameliorates Kras-induced MPN phenotypes and prolongs the survival of Kras mice. Our results that Sos1 acts as an oncogene are consistent with the identification of SOS1 gain-of-function mutations in pediatric myelodysplastic syndrome4,20 and other hematologic malignancies (supplemental Figure 5). Our study provides a rationale to target Sos1-oncogenic Kras interaction for treating oncogenic Kras-driven cancers.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors are grateful to Lawrence E. Samelson for providing the Sos1 fl/fl mice. The authors also thank the University of Wisconsin Carbone Comprehensive Cancer Center for use of its Shared Services (Flow Cytometry Laboratory, Genome Editing and Animal Models Shared Resource, and Experimental Pathology Laboratory) to complete this research. This work was supported by National Institutes of Health, National Cancer Institute R01 grant R01CA152108 and National Institutes of Health, National Heart, Lung, and Blood Institute R01 grant R01HL113066, and a Scholar Award from the Leukemia & Lymphoma Society (J.Z.). This work was also supported in part by National Institutes of Health/National Cancer Institute P30 CA014520-UW-Comprehensive Cancer Center Support.
Authorship
Contribution: X.Y. and J.Z. provided conception and design; X.Y. provided acquisition of data; X.Y., G.K., E.A.R., and J.Z. provided analysis and interpretation of data; X.Y., E.A.R., and J.Z. provided writing, review, and/or revision of the manuscript; G.K., Y.Z., and D.Y. provided technical or material support; and J.Z. provided study supervision.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Jing Zhang, Room 7453, WIMR II, McArdle Laboratory for Cancer Research, 1111 Highland Ave, University of Wisconsin-Madison, Madison, WI 53705; e-mail: zhang@oncology.wisc.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal