

Castleman disease (CD) describes a heterogeneous group of hematologic disorders that share characteristic lymph node histopathology. Patients of all ages present with either a solitary enlarged lymph node (unicentric CD) or multicentric lymphadenopathy (MCD) with systemic inflammation, cytopenias, and life-threatening multiple organ dysfunction resulting from a cytokine storm often driven by interleukin 6 (IL-6). Uncontrolled human herpesvirus-8 (HHV-8) infection causes approximately 50% of MCD cases, whereas the etiology is unknown in the remaining HHV-8-negative/idiopathic MCD cases (iMCD). The limited understanding of etiology, cell types, and signaling pathways involved in iMCD has slowed development of treatments and contributed to historically poor patient outcomes. Here, recent progress for diagnosing iMCD, characterizing etio-pathogenesis, and advancing treatments are reviewed. Several clinicopathological analyses provided the evidence base for the first-ever diagnostic criteria and revealed distinct clinical subtypes: thrombocytopenia, anasarca, fever, reticulin fibrosis/renal dysfunction, organomegaly (iMCD-TAFRO) or iMCD-not otherwise specified (iMCD-NOS), which are both observed all over the world. In 2014, the anti-IL-6 therapy siltuximab became the first iMCD treatment approved by the US Food and Drug Administration, on the basis of a 34% durable response rate; consensus guidelines recommend it as front-line therapy. Recent cytokine and proteomic profiling has revealed normal IL-6 levels in many patients with iMCD and potential alternative driver cytokines. Candidate novel genomic alterations, dysregulated cell types, and signaling pathways have also been identified as candidate therapeutic targets. RNA sequencing for viral transcripts did not reveal novel viruses, HHV-8, or other viruses pathologically associated with iMCD. Despite progress, iMCD remains poorly understood. Further efforts to elucidate etiology, pathogenesis, and treatment approaches, particularly for siltuximab-refractory patients, are needed.
Introduction
Castleman disease (CD) describes a group of heterogeneous hematologic disorders that share a spectrum of lymph node histopathology ranging from atrophic germinal centers with hypervascularization (hyaline vascular/hypervascular histopathological subtype) to hyperplastic germinal centers with polytypic plasmacytosis (plasmacytic histopathological subtype).1 Unicentric CD (UCD) involves a single region of enlarged lymph nodes with characteristic histopathology and relatively mild symptomatology, which can be cured with lymph node excision. In contrast, multicentric CD (MCD) involves systemic inflammation, multicentric lymphadenopathy with characteristic histopathology, cytopenias, and potentially fatal multiple organ dysfunction resulting from a cytokine storm often including interleukin 6 (IL-6).
From 42% to 67% of the 1569 to 1756 MCD cases diagnosed each year in the United States are caused by uncontrolled human herpesvirus-8 (HHV-8) infection.2,-4 In these HHV-8-associated MCD cases, HIV infection or, more rarely, another cause of immunosuppression enables HHV-8 to escape host immune control and signal for excessive cytokine production and polyclonal lymphoproliferation. Significant research attention on HHV-8-associated MCD has led to standardized treatment and improved patient outcomes. Rituximab is highly effective by depleting B cells, the primary HHV-8 reservoir; appropriate therapy leads to a 92% 5-year overall survival.5
The etiology in the half of MCD cases that are HIV-negative and HHV-8-negative is unknown. These patients, which can present at any age, are referred to as having idiopathic MCD (iMCD). Although the lymphoproliferation in iMCD is polyclonal, the appropriate disease classification for iMCD as an autoimmune disorder, autoinflammatory disorder, malignancy, or infectious disease is not known. iMCD has received significantly less research attention than HHV-8-associated MCD and is considerably less well understood. Until several years ago, the iMCD field lagged far behind many others in hematology (Table 1). Limited collaboration between researchers and no registries or biobanks to centralize data and biospecimens meant that studies were limited to small numbers of cases, if they were performed at all. There were no foundations focused on advancing iMCD research or engaging patients. Further limiting research, iMCD had not been rigorously defined within CD. Different classification systems (eg, HIV status, histopathological subtype, regions of lymphadenopathy) were used by some to subdivide CD; others lumped all cases of CD together. The lack of uniform subclassification caused confusion and limited comparison of studies that subdivided CD differently. There were also no diagnostic criteria, no treatment guidelines, and no unique ICD codes.
Progress made for iMCD research, 2012-2018
| State of iMCD research in 2012 | State of iMCD research in 2018 |
|---|---|
| Research coordination | |
| No physician, researcher, or patient communities | 400+ physicians and researchers connected through the CDCN and 2000+ patients connected virtually and through annual meetings |
| No registry or biorepository | ACCELERATE Natural History Registry and Biorepository are enrolling patients around the world via e-consent |
| Defining iMCD | |
| Different subclassification systems being used | Uniform classification system published in Blood1 |
| No diagnostic criteria for iMCD | International, evidence-based diagnostic criteria published in 201713 |
| Characterizing pathogenesis of iMCD and advancing treatment options | |
| Prevailing model of pathogenesis: an IL-6 secreting, lymph node “tumor” disorder | New model guiding research: complex cytokine storm disorder with an unknown etiology; several hypothesized etiologies under intense investigation |
| No genomic alterations identified in iMCD | First somatic mutation in iMCD published in 201820 ; other candidates are currently undergoing validation |
| No FDA-approved treatments for iMCD | Siltuximab became the first and only FDA-approved treatment of iMCD in 2014 |
| No treatment guidelines | International, evidence-based treatment guidelines in press39 |
| No drugs in development directed at any targets other than IL-6 | First clinical trial of relapsed/refractory iMCD with a drug directed at a target other than IL-6/IL-6R expected to begin enrollment in 2019 |
| State of iMCD research in 2012 | State of iMCD research in 2018 |
|---|---|
| Research coordination | |
| No physician, researcher, or patient communities | 400+ physicians and researchers connected through the CDCN and 2000+ patients connected virtually and through annual meetings |
| No registry or biorepository | ACCELERATE Natural History Registry and Biorepository are enrolling patients around the world via e-consent |
| Defining iMCD | |
| Different subclassification systems being used | Uniform classification system published in Blood1 |
| No diagnostic criteria for iMCD | International, evidence-based diagnostic criteria published in 201713 |
| Characterizing pathogenesis of iMCD and advancing treatment options | |
| Prevailing model of pathogenesis: an IL-6 secreting, lymph node “tumor” disorder | New model guiding research: complex cytokine storm disorder with an unknown etiology; several hypothesized etiologies under intense investigation |
| No genomic alterations identified in iMCD | First somatic mutation in iMCD published in 201820 ; other candidates are currently undergoing validation |
| No FDA-approved treatments for iMCD | Siltuximab became the first and only FDA-approved treatment of iMCD in 2014 |
| No treatment guidelines | International, evidence-based treatment guidelines in press39 |
| No drugs in development directed at any targets other than IL-6 | First clinical trial of relapsed/refractory iMCD with a drug directed at a target other than IL-6/IL-6R expected to begin enrollment in 2019 |
As a result, iMCD pathogenesis was poorly understood. Although IL-6 is the pathological driver in many cases of iMCD, the etiology, dysregulated cell types and signaling pathways, and other cytokines involved are unknown. Nevertheless, 2 monoclonal antibodies targeting IL-6 signaling were in development, but no other targets were being pursued. Four recent series of iMCD and HIV-negative/HHV-8-unknown MCD cases indicate a 50% to 77% 5-year overall survival.6,,-9 Poor understanding of pathogenesis has slowed new drug identification and contributed to its mortality. However, tremendous progress has been made during the last 6 years in defining iMCD, uncovering pathogenesis, and advancing treatment options, which will be reviewed in this article.
Defining and diagnosing iMCD
As a key early step to organizing the field in 2014, a uniform classification system was proposed on the basis of literature review and communication with experts. Patients with CD lymph node features should be divided first into UCD or MCD. Then, MCD should be further subdivided according to etiology into HHV-8-assoociated MCD (caused by HHV-8); polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, skin changes (POEMS)-associated MCD (caused by monoclonal plasma cells); iMCD (unknown etiology); and disorders mimicking MCD (Figure 1).1
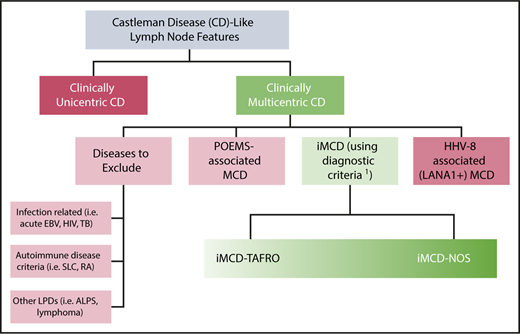
Uniform subclassification for patients with lymph nodes demonstrating features consistent with CD. Patients with lymph nodes demonstrating histology consistent with the CD spectrum (hypervascular/hyaline-vascular, plasmacytic, or mixed features) should be evaluated for number of regions of enlarged lymph nodes. If lymph node involvement is restricted to 1 site, the lesion most likely represents unicentric CD. If multiple sites are involved, patients should be evaluated for HHV-8, POEMS, and other infectious, malignant, and autoimmune disorders that can mimic iMCD. If these conditions are excluded, a diagnosis of iMCD should be considered. iMCD can be further subclassified into patients with iMCD with TAFRO syndrome (iMCD-TAFRO) and others whose subtype is not otherwise specified (iMCD-NOS). Adapted from Fajgenbaum et al.13
Uniform subclassification for patients with lymph nodes demonstrating features consistent with CD. Patients with lymph nodes demonstrating histology consistent with the CD spectrum (hypervascular/hyaline-vascular, plasmacytic, or mixed features) should be evaluated for number of regions of enlarged lymph nodes. If lymph node involvement is restricted to 1 site, the lesion most likely represents unicentric CD. If multiple sites are involved, patients should be evaluated for HHV-8, POEMS, and other infectious, malignant, and autoimmune disorders that can mimic iMCD. If these conditions are excluded, a diagnosis of iMCD should be considered. iMCD can be further subclassified into patients with iMCD with TAFRO syndrome (iMCD-TAFRO) and others whose subtype is not otherwise specified (iMCD-NOS). Adapted from Fajgenbaum et al.13
Once iMCD was clearly defined as an entity within CD, several systematic clinicopathological characterizations of iMCD were performed, including a review of 255 published cases2 and case series of 27, 31, and 44 patients with iMCD.10,-12 These descriptive studies provided key phenotypic data on iMCD and revealed that patients present with heterogenous clinical symptoms ranging from intense episodes of thrombocytopenia, anasarca, fever/elevated C-reactive protein (CRP), renal dysfunction/reticulin myelofibrosis, organomegaly, megakaryocytic hyperplasia, hypervascular or mixed lymph node histopathology, and normal gammaglobulin levels (iMCD-TAFRO) to a less intense inflammatory syndrome, normal/elevated platelet counts, plasmacytic or mixed lymph node histopathology, and polyclonal hypergammaglobulinemia whose subtype is not otherwise specified (iMCD-NOS).13 Therefore, iMCD should be further subdivided into iMCD-TAFRO and iMCD-NOS on the basis of clinical features. iMCD-TAFRO cases are most phenotypically similar to hemophagocytic lymphohistiocytosis and systemic lupus erythematosus, whereas iMCD-NOS cases are most similar to immunoglobulin G4-related disease, autoimmune lymphoproliferative syndrome, and Hodgkin lymphoma. Although iMCD-TAFRO was first described in Japan, both clinical subtypes are observed worldwide.
In 2015, an international working group of 34 experts from 8 countries on 5 continents established international consensus diagnostic criteria for iMCD, based on evidence from 244 clinical cases and 88 tissue samples. Diagnosis of iMCD requires both major criteria (multicentric lymphadenopathy and biopsy-proven histopathology on the iMCD spectrum), at least 2 of 11 minor criteria with at least 1 laboratory abnormality and exclusion of infectious, malignant, and autoimmune disorders that can mimic iMCD (eg, acute Epstein-Barr virus, lymphoma, systemic lupus erythematosus). The spectrum of iMCD lymph node histopathology includes a constellation of hyperplastic or regressed germinal centers, often with widened mantle zones in an onion-skin appearance, prominent follicular dendritic cells (FDCs) occasionally appearing dysplastic, hypervascularization, and polytypic plasmacytosis. Staining for HHV-8 must be negative by latency-associated nuclear antigen 1. Minor criteria include elevated CRP or erythrocyte sedimentation rate, anemia, thrombocytopenia or thrombocytosis, hypoalbuminemia, renal dysfunction or proteinuria, polyclonal hypergammaglobulinemia, constitutional symptoms, hepatosplenomegaly, effusions or edema, cherry hemangiomata or violaceous papules, and lymphocytic interstitial pneumonitis. Guidelines recommend against using IL-6 levels for diagnosis because of a lack of sensitivity or specificity.13 Reflecting improvements in its classification, a unique ICD-10 code was established for CD (D47.Z2) in 2016.
Characterizing pathogenesis of iMCD
The prevailing model of iMCD pathogenesis in the literature before 2014 was that the enlarged lymph nodes were tumors that produced IL-6, which in turn led to systemic inflammation and organ dysfunction. After a literature review and discussion among experts, a new conceptual framework was proposed whereby the enlarged lymph nodes and histopathological features are reactive changes to the elevated IL-6 and/or other circulating factors in the cytokine storm. The heterogeneity of iMCD and overlap with neoplastic, infectious, and rheumatologic disorders suggest that multiple processes, each involving immune dysregulation and elevated cytokine release, may be able to give rise to iMCD. Four etiologies were proposed, as well as candidate dysregulated cell types, signaling pathways, and driver cytokines, as a framework to guide hypothesis-driven iMCD research.1 Although many questions remain, progress has been made, which is described here and included in the updated model of pathogenesis (Figure 2).
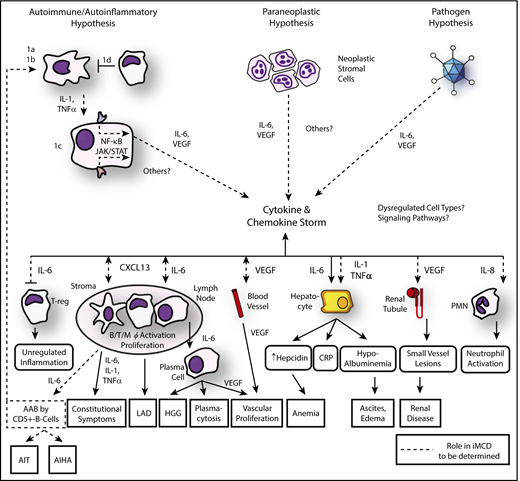
Updated model of iMCD pathogenesis. Three hypothesized mechanisms may be responsible for the iMCD cytokine and chemokine storm: first, the autoimmune/autoinflammatory hypothesis involves (1a) auto-antibodies triggering proinflammatory cytokine release by antigen-presenting cells that induce the as-yet-unknown hypercytokine-secreting cell to release IL-6 or other pathologic cytokines; (1b/c) dysregulated signaling in an antigen presenting cell or other as-yet-unknown hypercytokine-secreting cell releasing IL-6 or other pathologic cytokines, or (1d) a defect in the regulation of activated inflammatory cells. The cytokine and chemokine storm is perpetuated by positive feedback of IL-6, other pathologic cytokines, and/or possibly further auto-antibody stimulation. Second, the paraneoplastic syndrome hypothesis involves a somatic mutation in benign or malignant cells inside or outside of the lymph node that causes constitutive cytokine release. Preliminary data suggest these may be lymph node stromal cells. Third, the pathogen hypothesis involves either infection with HHV-8 that is clinically undetectable, a novel virus, or another pathogen signaling proinflammatory cytokines. An active infection by a single virus is less likely based on preliminary data generated from pathogen discovery studies. Regardless of the etiology, the cytokine and chemokine storm is the common pathway that results in the subsequent clinical and histopathological features of iMCD. AAB, autoantibodies; AIHA, autoimmune hemolytic anemia; AIT, autoimmune thrombocytopenia; LAD, lymphadenopathy; PMN, polymorphic neutrophil. Adapted from Fajgenbaum et al.1
Updated model of iMCD pathogenesis. Three hypothesized mechanisms may be responsible for the iMCD cytokine and chemokine storm: first, the autoimmune/autoinflammatory hypothesis involves (1a) auto-antibodies triggering proinflammatory cytokine release by antigen-presenting cells that induce the as-yet-unknown hypercytokine-secreting cell to release IL-6 or other pathologic cytokines; (1b/c) dysregulated signaling in an antigen presenting cell or other as-yet-unknown hypercytokine-secreting cell releasing IL-6 or other pathologic cytokines, or (1d) a defect in the regulation of activated inflammatory cells. The cytokine and chemokine storm is perpetuated by positive feedback of IL-6, other pathologic cytokines, and/or possibly further auto-antibody stimulation. Second, the paraneoplastic syndrome hypothesis involves a somatic mutation in benign or malignant cells inside or outside of the lymph node that causes constitutive cytokine release. Preliminary data suggest these may be lymph node stromal cells. Third, the pathogen hypothesis involves either infection with HHV-8 that is clinically undetectable, a novel virus, or another pathogen signaling proinflammatory cytokines. An active infection by a single virus is less likely based on preliminary data generated from pathogen discovery studies. Regardless of the etiology, the cytokine and chemokine storm is the common pathway that results in the subsequent clinical and histopathological features of iMCD. AAB, autoantibodies; AIHA, autoimmune hemolytic anemia; AIT, autoimmune thrombocytopenia; LAD, lymphadenopathy; PMN, polymorphic neutrophil. Adapted from Fajgenbaum et al.1
Etiology
The cytokine storm that drives iMCD is hypothesized to be caused by an uncontrolled infection (pathogen hypothesis), auto-antibodies or auto-reactive T cells associated with predisposing germline mutations (autoimmune hypothesis), germline mutations in genes regulating inflammation (autoinflammatory hypothesis), and/or somatic mutations in monoclonal lymph node cells that lead to ectopic cytokine secretion (paraneoplastic hypothesis).
Pathogen hypothesis.
A pathogen was considered the most likely etiological mechanism for iMCD, based on its similarities with HHV-8-associated MCD, its episodic course potentially reflecting a latent-lytic viral cycle, and the numerous lymphotrophic viruses similar to HHV-8. Even though iMCD is, by definition, HHV-8 negative by latency-associated nuclear antigen 1 staining, many in the field hypothesized that iMCD is caused by HHV-8 that is falsely not detected by clinical assays (eg, because of viral epitope mutation). Others proposed that an undiscovered, novel virus with homology to HHV-8 may drive iMCD. To test the pathogen hypothesis, a multinational collaboration between 7 institutions was established to perform VirCapSeq,14 a positive-selection, high-throughput, hybrid-capture RNA sequencing approach for detecting novel and known viruses, on tissue from 11 patients with iMCD, 12 patients with UCD, 2 patients with HHV-8-associated MCD, and 6 lymphoma control patients. The results did not support the 2 hypotheses (eg, HHV-8 causes iMCD, infection with a novel virus causes iMCD) we set out to investigate. Consistent with prior clinical testing, HHV-8 was not detected in any iMCD, UCD, or lymphoma cases; however, HHV-8 was detected in both HHV-8-associated MCD cases as expected. No novel viruses were discovered, but Herpesviridae, including Epstein-Barr virus, HHV-6 and HHV-7, and non-Herpesviridae were detected inconsistently across the iMCD cases.15 Whether these infections are pathologic, contributory to clinical severity, coincidental, or secondary to iMCD immune dysregulation remains to be determined. To further test the pathogen hypothesis, CD samples are being analyzed using an orthogonal method that detects nucleotide sequences from all sequenced viruses, as well as human pathogenic bacteria, fungi, and parasites.
Autoimmune and autoinflammatory hypotheses.
Autoimmune and autoinflammatory mechanisms were proposed as etiological drivers of iMCD, based on its clinicopathological overlap with conditions such as systemic lupus erythematosus and hemophagocytic lymphohistiocytosis. A recent review found that 30% of published iMCD cases have autoimmune hemolytic anemia or auto-antibodies.2 These auto-antibodies may initiate immune activation and cytokine production, or they may be byproducts of other etiologies. Immune repertoire profiling is underway to assess clonality and search for self-reactive T and B cells. Further research is needed, including screening of sera for auto-antibodies, which is planned to begin in 2019.
A number of genomic sequencing studies have recently been published or are currently being performed to search for germline mutations that may predispose patients with iMCD to autoimmunity or cause inflammatory dysregulation. Genomic sequencing of DNA, believed to represent constitutional DNA, has identified mutations in iMCD patients that are associated with monogenic inflammatory disorders, such as CECR1 in deficiency of ADA2 (DADA2) and MEFV in familial Mediterranean fever.16,17 These cases and others with autoinflammatory disorders can demonstrate clinical features and histopathology that overlap with iMCD. The clinical features that led to the diagnosis of iMCD in these patients are likely caused by underlying DADA2 or familial Mediterranean fever, which should be the focus of treatment. Additional research is needed to determine whether these disorders should be considered exclusionary to iMCD. Interestingly, Oksenhendler at al recently reported 3 patients with iMCD born from consanguineous parents.11 Further, 4 families were recently identified that each had 2 patients with CD in them. Sequencing of these families is currently in process.
Paraneoplastic hypothesis.
The paraneoplastic hypothesis was suggested on the basis of iMCD’s overlapping histopathological features with Hodgkin lymphoma, which involves a small population of somatically mutated, monoclonal cells driving excessive cytokine production and reactive lymphoproliferation. Patients with iMCD have an increased rate of malignancies compared with age-matched control patients, possibly indicating that a premalignant clone responsible for iMCD goes on to acquire additional mutations that lead to cancer.2 Limited data exist in support of this hypothesis. Chang et al found monoclonal cells in 4 of 4 iMCD lymph nodes. As the lymphocytes were polyclonal in the 4 cases, which is typical in iMCD, the authors proposed that the monoclonal cells were likely lymph node stromal cells.18 A somatic translocation [46,XY,t(7;14)(p22;q22)] at the IL-6 locus was found in 1993 in an HIV-negative/HHV-8-unknown MCD patient’s lymph node.19 More recently, a next-generation sequencing tumor panel of 405 genes identified the first somatic mutation in lymph node tissue from a patient with iMCD, a missense mutation in DNMT3A (L295Q). The mutation was not detected in the patient’s peripheral blood, bone marrow, or adipose tissue, suggesting that this somatic mutation does not represent clonal hematopoiesis of indeterminate potential. Copy number variants in ETS, PTPN6, TGFBR2, and TUSC3 were also reported.20 Of note, no somatic mutations were identified in lymph node tissue from the other 2 iMCD cases sequenced in this study. Investigations into the cell harboring the DNMT3A mutation and the potential contribution of DNMT3A to iMCD pathogenesis are in process. A somatic mutation in MAP2K2 was identified in lymph node tissue from another patient with iMCD by a separate 400-gene tumor panel, which is currently undergoing functional validation (personal communications: Wenbin Xiao, Memorial Sloan Kettering Cancer Center). Whole-exome sequencing is underway on 30 iMCD lymph nodes, which should provide further insights into somatic mutations in iMCD.
Driver cytokines
Although the etiology is unknown, increased IL-6 signaling is the established driver of iMCD symptomatology and pathogenesis in a subset of patients. IL-6 is a multifunctional cytokine that induces B-cell and plasma cell maturation, acute inflammation, and secretion of vascular endothelial growth factor (VEGF). Intensity of iMCD symptoms is significantly correlated with IL-6 levels, which can be highly elevated during flares.21 Further, IL-6 overexpressing mice recapitulate many features of iMCD, which are abrogated with IL-6 neutralization. Moreover, administration of IL-6 to humans leads to an iMCD-like syndrome.22 As a result, monoclonal antibodies directed at IL-6 (siltuximab) and the IL-6 receptor (tocilizumab) were developed for iMCD.
Tocilizumab was approved for iMCD in Japan in 2005 on the basis of a single open-label prospective study (N = 35) demonstrating improvements in constitutional symptoms, laboratory markers, and lymphadenopathy with few adverse events.23 However, overall response criteria were never assessed, and it was not approved for iMCD outside of Japan because of the lack of randomized controlled trial data. In 2014, siltuximab was approved for iMCD in the United States and many other countries on the basis of a 34% durable response rate (compared with 0% in the placebo group) in a phase 2 randomized, double-blind, placebo-controlled trial (n = 79) as well as positive data from a phase 1 trial (n = 34) and long-term safety study (n = 19).24,-26 Although anti-IL-6 therapy is highly effective for many patients, two-thirds of patients receiving siltuximab in the phase 2 trial did not meet response criteria, approximately half of which did not have elevated IL-6.21 Thus, there are likely to be other cytokines or pathways that can drive iMCD pathogenesis.
Until recently, systematic serum proteomic or cytokine quantification had never been performed in iMCD. Iwaki et al measured 18 cytokines during flare in 11 patients with iMCD-TAFRO and 5 patients with iMCD-NOS compared with 21 healthy control patients.27 Surprisingly, IL-6 levels were not statistically different between the 3 groups; median IL-6 was 0 pg/mL for all 3 groups. Among the 17 other cytokines whose levels were assayed, IL-10, IL-23, and VEGF were significantly elevated in both iMCD groups compared with healthy control patients, and the chemokine CXCL10 was significantly elevated in iMCD-TAFRO compared with both iMCD-NOS and healthy control patients.27 Evidence has also accumulated implicating VEGF, a potent angiogenic factor, in iMCD pathogenesis. VEGF was elevated in 16/20 published iMCD cases reporting VEGF levels, and they often parallel disease activity.2 This may explain the eruptive cherry hemangiomatosis, capillary leak syndrome, and lymph node hypervascularity that can be observed in iMCD.13,28
A systematic proteomic analysis of 1129 analytes in paired flare-remission plasma samples from 6 patients with iMCD was also recently performed. This study revealed that cytokine and chemokine signaling were the most enriched upregulated pathways. Chemokines, including several that are often produced by lymph node stromal cells, were significantly more upregulated than interleukins and other cytokines; “chemokine storm” was proposed to describe these observations. CXCL13 was the most upregulated cytokine during flare across all patients. Further, its expression was significantly increased in iMCD lymph node germinal centers compared with controls in a mesh-like pattern, possibly representing FDCs.29 CXCL13 is primarily produced by FDCs to home B cells into germinal centers for selection and maturation into plasma cells and to maintain appropriate germinal center morphology. Given the plasmacytosis and dysmorphic germinal centers characteristic of iMCD, dysregulation of CXCL13 may be important to iMCD pathogenesis. To follow-up on these promising proteomic findings, an international, 8-party collaborative study was established to quantify 1300 serum analytes in 362 samples from 100 patients with iMCD and 100 control patients (20 HHV-8-associated MCD, 20 rheumatoid arthritis, 20 Hodgkin lymphoma, and 40 healthy control patients). This study, which will be completed in 2018, aims to identify candidate biomarkers for diagnosis and response to siltuximab and generate hypotheses regarding signaling pathways and driver cytokines responsible for iMCD.
Although IL-1β was not elevated in the Iwaki et al27 or Pierson et al29 studies, administration of an IL-1 receptor antagonist has been effective in a few cases, suggesting a potential pathological role for IL-1β, which can lead to IL-6 production through nuclear factor (NF)-κB activation.30,31 Inhibition of VEGF, CXCL13, or other candidate driver cytokines has not been reported in iMCD; future efforts to do so may shed light on their roles.
Dysregulated cell types
Limited research to date has generated conflicting reports regarding the cell types responsible for driving iMCD pathogenesis. Candidates include plasma cells, B cells, T cells, macrophages, and follicular dendritic cells.32,33 Given the heterogeneous spectrum of histopathology, symptomatology, and treatment response, different cell types may be involved in different subgroups.
Evidence for a pathogenic role of B cells and plasma cells comes from patients with sheet-like plasmacytosis and hypergammaglobulinemia, who respond to B-cell depletion with rituximab or proteasome inhibitors targeting plasma cells. However, many patients, particularly those with iMCD-TAFRO, do not demonstrate these features or respond to rituximab.2
Recent findings have implicated activated T cells as a potential pathogenic driver in iMCD. Elevated serum soluble IL-2 receptor (sIL2R), which is shed by activated T cells, was found in 20/21 published cases reporting sIL2R levels.2 Increased immature TdT+ T cells are observed in iMCD, possibly representing general dysregulation of T cells.34 Furthermore, the T-cell immunosuppressants cyclosporine and sirolimus have been effective in iMCD-TAFRO cases.2
Evidence implicating lymph node stromal cells, particularly FDCs, in iMCD pathogenesis is discussed here: Chang et al18 proposed that stromal cells made up the monoclonal lymph node cells, Pierson et al29 found that chemokines primarily produced by stromal cells are highly upregulated in plasma during flare, and FDCs can be abnormally prominent or dysplastic in appearance in iMCD.13 Immunophenotyping of peripheral blood and lymph node tissue by flow cytometry, as well as identification of cytokine-secreting cells with in situ hybridization, are in process.
Signaling pathways
Although it is not known which intracellular signaling pathways are involved in iMCD, candidate pathways include those upstream and downstream of IL-6. NFκB signaling is the primary transcription factor involved in IL-6 production. Although no experimental work has been done to characterize NFκB signaling in iMCD, bortezomib, a proteasome inhibitor believed to inhibit NFκB, has induced a few responses.35,36 Janus kinase (JAK)/signal transducer activator of transcription 3 (STAT3), mitogen-activated protein kinase (MAPK), and phosphatidylinositol-3-kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR) are the 3 primary pathways activated by IL-6. Knocking out CCAAT/enhancer binding protein b (C/EBPb) causes an iMCD-like phenotype in mice because of unopposed STAT3.37 Pierson et al found that MAPK and PI3K/Akt/mTOR signaling are both enriched among the most up- and downregulated plasma proteins during iMCD flare, and the greatest number of compounds (6 of the top 20) that downregulate expression (in vitro) of the most upregulated proteins in iMCD-TAFRO patients, including VEGF, CXCL13, and other chemokines, target PI3K and/or mTOR.29 One treatment-refractory patient with iMCD demonstrating increased mTOR signaling, elevated VEGF, and T-cell activation has experienced a prolonged remission on the mTOR inhibitor sirolimus.38 We have also discovered significantly increased mTOR activation in lymph node tissue from patients with iMCD compared with reactive controls (D.C.F. and Dustin Shilling, unpublished data). On the basis of these preliminary data, a clinical trial of sirolimus in anti-IL6-refractory iMCD is being planned. This will be the first clinical trial in iMCD targeting a pathway other than IL-6/IL-6 receptor signaling. Further research is needed to identify the precise cell types in which these signaling pathways are dysregulated.
Advancing treatment options
A working group of 42 international experts recently established the first-ever evidence-based consensus treatment guidelines for iMCD based on review of 344 cases and expert opinion.39 The working group recommends siltuximab (11 mg/kg every 3 weeks) ± corticosteroids as first-line therapy for all patients, based on effectiveness, safety profile, worldwide approvals, and rigorous study methodology. Treatment response should be evaluated by symptomatic (fatigue, anorexia, fever, weight change), biochemical (CRP, hemoglobin, albumin, estimated glomerular filtration rate), and radiologic criteria (computed tomography ± positron emission tomography). Patients who respond to siltuximab should taper off corticosteroids and continue receiving siltuximab monotherapy indefinitely. Treatment of patients who do not respond to siltuximab should be tailored by disease severity into nonsevere or severe (intensive care, progressive organ dysfunction [hepatic, renal, cardiac, pulmonary]). Patients with nonsevere disease (no intensive care or progressive organ dysfunction) who fail to adequately respond to siltuximab after 3 to 4 doses should receive rituximab (375 mg/m2 × 4-8 weekly doses) ± corticosteroids ± an immunomodulatory/immunosuppressive agent, such as cyclosporine, sirolimus, anakinra, thalidomide, or bortezomib. Among responders, patients with a mild pretreatment disease course may be carefully observed off of treatment, whereas patients with a more intense pretreatment disease course should be maintained on an immunomodulatory/immunosuppressive agent. Third-line treatment of patients with nonsevere disease that fail to respond to rituximab after 4 to 8 weekly doses involves an immunomodulatory/immunosuppressive agent. For patients with severe disease, accelerated weekly dosing of siltuximab with high-dose corticosteroids (eg, methylprednisolone 500 mg daily) is recommended. Any sign of worsening organ function should immediately trigger the initiation of chemotherapy (eg, rituximab-cyclophosphamide-doxorubicin-vincristine-prednisone, bortezomib-dexamethasone-thalidomide-adriamycin-cyclophosphamide-etoposide-rituximab, cyclophosphamide-etoposide-rituximab) to ablate the hyperactivated immune system and stem the cytokine storm. We recommend additional rounds of combination chemotherapy with or without immunomodulators/immunosuppressants if insufficient response is achieved. Patients with iMCD in the intensive care unit can have dramatic and durable turnarounds if given the right agents, so persistent, aggressive treatment is recommended.
Given the large proportion of patients with iMCD who do not respond to siltuximab, discovery of predictive biomarkers of response could help to personalize treatment beyond disease severity. We recently analyzed 38 pretreatment laboratory parameters from the phase 2 trial of siltuximab in patients with iMCD meeting criteria for treatment response or failure to develop a predictive model of response. Univariate analyses identified 8 pretreatment parameters significantly different between treatment responders and failures: albumin, CRP, immunoglobulin G, immunoglobulin A, fibrinogen, hemoglobin, sodium, and triglycerides. Of note, pretreatment IL-6 is not significantly associated with response to siltuximab. Stepwise logistic regression analysis of these candidate parameters identified a top-performing model that included fibrinogen, immunoglobulin G, hemoglobin, and CRP, suggesting that patients with an inflammatory syndrome are the best candidates for siltuximab. Although the model accurately discriminated treatment responders from failures (area under curve, 0.86; 95% confidence interval, 0.73-0.95), further validation is needed in a separate cohort before this model is adopted in clinical practice.40
A lack of robust, centralized data and biospecimens has slowed the discovery of novel treatments and predictive biomarkers of response needed to achieve personalized medicine in iMCD. To overcome this hurdle, the ACCELERATE natural history registry was launched in 2016 in partnership between the Castleman Disease Collaborative Network (CDCN), University of Pennsylvania’s Penn Castleman Disease Center, and Janssen Pharmaceutica NV.41 The goals are to leverage real-world data to generate clinical insights, validate the diagnostic criteria, identify personalized treatment approaches based on clinical phenotype, and promote research by making the data available to the community. Patients anywhere in the world can e-consent themselves directly online (www.CDCN.org/accelerate) for medical record acquisition and extraction by trained data analysts at the Penn Castleman Disease Center. All cases are reviewed by an expert panel to grade the likelihood of the patient having CD and assessed for treatment response. In the first 1.5 years, 156 patients have enrolled into ACCELERATE. Approximately 3000 data elements have been extracted for each patient fully entered. We plan to continue to enroll approximately 100 patients per year.
Future directions
Significant progress toward improving iMCD patient outcomes has occurred over the last several years since the creation of the CDCN in 2012 (Table 1). The CDCN has spearheaded a “collaborative network approach,” described in Figure 3, to overcome the aforementioned hurdles and accelerate CD research and treatment discovery.42 However, a great deal of work remains to improve understanding of pathogenesis and discover novel treatment approaches, particularly for patients who fail siltuximab. With the CDCN’s collaborative network, international research agenda, and research infrastructure such as ACCELERATE in place, the potential for improving understanding of iMCD, discovering new treatment options, and improving outcomes for patients like me is great.
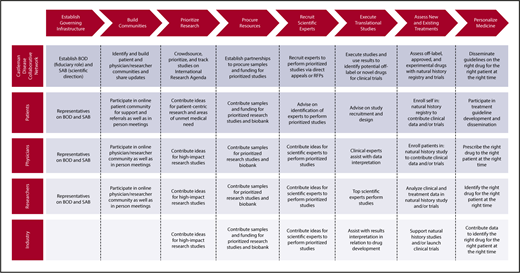
The Collaborative Network Approach being used to advance research for iMCD. The Castleman Disease Collaborative Network, patients, physicians, researchers, and industry are taking a systematic approach to advance CD research that may be repeatable by other disease fields. BOD, board of directors; RFP, request for proposals; SAB, scientific advisory board.
The Collaborative Network Approach being used to advance research for iMCD. The Castleman Disease Collaborative Network, patients, physicians, researchers, and industry are taking a systematic approach to advance CD research that may be repeatable by other disease fields. BOD, board of directors; RFP, request for proposals; SAB, scientific advisory board.
Acknowledgments
The author thanks Dustin Shilling for review and insights; Arthur Rubenstein for guidance; and Helen Partridge, Frits van Rhee, Jason Ruth, Chris Nabel, Sheila Pierson, Mary Zuccato, and members of the CDCN Scientific Advisory Board for contributions to the iMCD field.
This work was supported, in part, by National Heart, Lung, & Blood Institute of the National Institutes of Health Grant R01HL141408.
Authorship
Contribution: D.C.F. wrote the manuscript.
Conflict of interest disclosure: D.C.F. has received research funding from Janssen Pharmaceuticals and serves on the Board of Directors for the Castleman Disease Collaborative Network. Off-label drug use: None disclosed.
Correspondence: David Fajgenbaum, Hospital of the University of Pennsylvania, Division of Translational Medicine & Human Genetics, 3400 Spruce St, Silverstein 5, Suite 5100, Philadelphia, PA 19104; e-mail: davidfa@pennmedicine.upenn.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal