

Key Points
Acenocoumarol is the most efficient oral anticoagulant with the least variation of resistance among naturally occurring VKOR mutations.
Warfarin and fluindione are equipotent for anticoagulation control but have different mechanisms of action on VKOR inactivation.

Abstract
Warfarin, acenocoumarol, phenprocoumon, and fluindione are commonly prescribed oral anticoagulants for the prevention and treatment of thromboembolic disorders. These anticoagulants function by impairing the biosynthesis of active vitamin K–dependent coagulation factors through the inhibition of vitamin K epoxide reductase (VKOR). Genetic variations in VKOR have been closely associated with the resistant phenotype of oral anticoagulation therapy. However, the relative efficacy of these anticoagulants, their mechanisms of action, and their resistance variations among naturally occurring VKOR mutations remain elusive. Here, we explored these questions using our recently established cell-based VKOR activity assay with the endogenous VKOR function ablated. Our results show that the efficacy of these anticoagulants on VKOR inactivation, from most to least, is: acenocoumarol > phenprocoumon > warfarin > fluindione. This is consistent with their effective clinical dosages for stable anticoagulation control. Cell-based functional studies of how each of the 27 naturally occurring VKOR mutations responds to these 4 oral anticoagulants indicate that phenprocoumon has the largest resistance variation (up to 199-fold), whereas the resistance of acenocoumarol varies the least (<14-fold). Cell-based kinetics studies show that fluindione appears to be a competitive inhibitor of VKOR, whereas warfarin is likely to be a mixed-type inhibitor of VKOR. The anticoagulation effect of these oral anticoagulants can be reversed by the administration of a high dose of vitamin K, apparently due to the existence of a different enzyme that can directly reduce vitamin K. These findings provide new insights into the selection of oral anticoagulants, their effective dosage management, and their mechanisms of anticoagulation.
Introduction
Fluindione and coumarin derivatives (such as warfarin, acenocoumarol, and phenprocoumon) are known as vitamin K antagonists (VKAs), and are widely used oral anticoagulants in the prevention and treatment of thromboembolic disorders.1,2 Although warfarin is the most commonly used VKA worldwide, in some countries, other VKAs are more often prescribed.3,4 These drugs exert their anticoagulant effects by impairing the biosynthesis of functional vitamin K–dependent clotting factors through the inhibition of vitamin K epoxide reductase (VKOR) activity. VKOR is responsible for the regeneration of the reduced form of vitamin K (vitamin K hydroquinone [KH2]), an essential cofactor for the posttranslational carboxylation of several clotting factors.5 Inadequate KH2 results in the production of undercarboxylated and/or noncarboxylated forms of coagulation factors with impaired biological activities. The anticoagulation efficacy of VKAs is evaluated by the prothrombin time and the international normalized ratio (INR). A beneficial therapeutic INR range is between 2.0 and 3.0, with a lower or a higher INR increasing the risk of thromboembolic or hemorrhagic events, respectively.6 Therefore, management of a therapeutic INR with oral anticoagulants is challenging due to the narrow therapeutic index and the broad individual patient variability of VKA-dosing requirements.7,8 Despite these well-known drawbacks and the development of novel oral anticoagulants over the past decade,7 VKAs, such as warfarin, are still the most commonly prescribed anticoagulants globally.1,9,10
VKA dosage requirements are influenced by multiple factors. These include individual patients, variable vitamin K diet intakes, food and drug interactions, and genetic variations of the VKA target and metabolic enzymes (VKOR and CYP2C9, cytochrome P450 2C9).11 Genotypes of the VKOR and CYP2C9 genes have been strongly associated with VKA dosage requirements.12 Several pharmacogenetic dosing algorithms have been proposed to assist physicians in estimating appropriate VKA dosages.13-16 VKOR pharmacogenetics is thought to be so clinically useful that the US Food and Drug Administration revised warfarin product labels to include the genotypes of the VKOR gene in warfarin dosage recommendations.17 It has been shown that ∼30% of patients receiving warfarin would benefit from VKOR pharmacogenetics at the beginning of their warfarin therapy.18,19 However, controversial results exist on the usefulness of the genotype-guided VKA dosage control.20,21
Currently, VKOR pharmacogenetics only takes into account single-nucleotide polymorphisms in the noncoding region of the VKOR gene. The most commonly used polymorphism in VKOR pharmacogenetics is c.-1639G>A (rs9923231), a mutation in the promoter region of VKOR believed to be the causative mutation for a low-dose VKA requirement.22,23 Although the c.1173C>T polymorphism found in intron 1 is also associated with a low-dose warfarin phenotype,24,25 and the 3′ untranslated region polymorphism of c.3730G>A (rs7294) appears to be associated with a high-dose warfarin phenotype,25 according to the 2017 updated guideline for pharmacogenetics-guided warfarin dosing from the Clinical Pharmacogenetics Implementation Consortium, the c.-1639G>A polymorphism is the only variant strongly associated with warfarin dosage.26 Nevertheless, a combination of the pharmacogenetics of VKOR and CYP2C9, as well as the clinical variables, can only explain up to 50% of the clinical warfarin dosage variabilities.27 Therefore, it would be potentially beneficial to include the missense mutations identified in the VKOR-coding region for VKA dosage management, as these mutations often result in VKOR molecules that are more resistant to VKA inhibition.28,29
There are ∼40 missense mutations identified in the coding region of VKOR that cause the VKA-resistant phenotype in rats, mice, and humans.28-30 However, it is not clear how each of these naturally occurring mutations affect the binding of VKOR to its substrate (vitamin K epoxide [KO]) and/or its inhibitors (such as VKAs). Additionally, controversial results were obtained for the correlation between the clinical VKA-resistant phenotypes and the functional studies of the VKA-resistant VKOR mutations.31-33 Functional studies of these mutations, using the conventional in vitro dithiothreitol-driven VKOR activity assay, suggest that VKOR mutations detected in patients resistant to VKA therapy are not all associated with VKA-resistant VKOR activity.31 This result, in general, agrees with our cell-based functional study of VKOR mutations.32 However, results from a similar cell-based study suggest that warfarin resistance of all naturally occurring VKOR mutations corresponds well to the patient’s oral anticoagulant-resistant phenotype.33 Therefore, a better understanding of the correlation between VKOR mutations and the resistance of VKAs is required. This is not only important for the management of VKA dosages, but also important for understanding the molecular mechanism of VKAs targeting VKOR.
In this study, we compared the efficacy of the 4 clinically used VKAs in the inhibition of VKOR activity and examined the resistance variations of these VKAs among the naturally occurring VKOR mutations, using our recently established double-gene knockout (DGKO) cell-based VKOR activity assay.32 Additionally, we performed cell-based enzyme kinetics studies to explore the possible mechanistic differences of inactivation of VKOR by these VKAs. Results from this study will provide new insights into the management of VKAs and their mechanisms of anticoagulation.
Methods
Materials and cell lines
Acenocoumarol, phenprocoumon, and fluindione were purchased from Santa Cruz Biotechnology, Inc (Dallas, TX). Warfarin, vitamin K1, and KO were obtained from Sigma-Aldrich (St. Louis, MO). Xfect transfection reagent was from Clontech Laboratories, Inc (Mountain View, CA). Mammalian dual expression vector pBudCE4.1, mammalian cell culture medium, and oligonucleotide primers were from Life Technologies Corp (San Diego, CA). The QuickChange site-directed mutagenesis kit was from Agilent Technologies, Inc (Santa Clara, CA). Mouse anti-carboxylated factor IX gla domain monoclonal antibody was from Green Mountain Antibodies (Burlington, VT). Horseradish peroxidase–conjugated affinity-purified sheep anti-human protein C was from Affinity Biologicals, Inc (Ancaster, ON, Canada). ABTS (2,2′-Azino-bis (3-ethylbenzothiazoline-6-sulfonic acid) diammonium salt) peroxidase substrate kit for enzyme-linked immunosorbent assay (ELISA) was from KPL Inc (Gaithersburg, MD).
The human embryonic kidney 293 (HEK293) cell line was from ATCC (Manassas, VA). HEK293 cells stably expressing FIXgla-PC as the reporter protein (FIXgla-PC/HEK293) were obtained as previously described.34 FIXgla-PC/HEK293 cells with their endogenous VKOR and VKOR-like enzyme knocked out (DGKO) were obtained by transcription activator-like effector nuclease–mediated genome editing, as previously described.32
DNA manipulations and plasmids construction
Mammalian dual-expression vector pBudCE4.1, with Metridia luciferase complementary DNA (cDNA; as internal control for transfection efficiency) cloned into one of the multicloning sites (pBudCE4.1-Met.Luc), was used as the cloning and expression vector for expressing VKOR and its variants, as previously described.35 The cDNA of wild-type VKOR was cloned in pBudCE4.1-Met.Luc by restriction sites PstI and XbaI. All naturally occurring VKOR mutations were created by a QuickChange mutagenesis kit using the wild-type VKOR construct as the template. The nucleotide sequences of all the constructs were verified by DNA sequencing at Eton Bioscience Inc (Research Triangle Park, NC).
Cell-based functional study of VKOR and its mutants
Functional studies of VKOR and its mutants were performed in DGKO FIXgla-PC/HEK293 reporter cells, as previously described.32 This assay is based on the ability of the exogenously expressed VKOR or its mutants to convert KO to vitamin K in HEK293 cells to support vitamin K–dependent carboxylation of the reporter protein FIXgla-PC. Briefly, plasmid DNA of pBudCE4.1 containing the cDNA of wild-type or mutant VKOR was transiently expressed in the DGKO FIXgla-PC/HEK293 cells using Xfect transfection reagent. At 5 hours posttransfection, the transfection medium was replaced by a complete medium containing 5 µM KO for activity assay. For the VKA-resistance study, the transfection medium was replaced by complete medium containing 5 µM KO with increasing concentrations of VKAs. For the KO-binding study, the transfection medium was replaced by complete medium containing increasing concentrations of KO. After incubation for 48 hours, the cell culture medium was collected and directly used for ELISA to determine the level of carboxylated reporter protein, FIXgla-PC. VKOR activity was expressed as normalized carboxylated FIXgla-PC. Wild-type VKOR activity was normalized to 100%. The VKA resistance was evaluated by determining the half-maximal inhibition concentration (IC50) of the VKAs using GraphPad software. The KO-binding affinity was evaluated by determining the half-maximal stimulation concentration of KO (EC50; or apparent Michaelis-Menten constant [Km]).
Cell-based enzyme kinetics study on VKOR
All of the cell-based enzyme kinetics studies were performed in FIXgla-PC/HEK293 reporter cells. Briefly, FIXgla-PC/HEK293 cells were seeded in a multiwell cell culture plate 1 day prior to the substrate/inhibitor treatment. On the following day, the cell culture medium was replaced with complete medium containing increasing concentrations of KO with or without a certain concentration of VKAs, as indicated in the figure legends. After incubation for 24 hours, the cell culture medium was harvested and used directly to measure the levels of carboxylated FIXgla-PC. Experimental data were used to determine the inhibition model by the Lineweaver-Burk plot, Cornish-Bowden plot, and GraphPad software.
Results
Inhibition of VKOR activity by clinically used VKAs
The most frequently used VKAs are 4-hydroxycoumarin derivatives, including warfarin, acenocoumarol, and phenprocoumon1 (Figure 1A). However, fluindione, a derivative of 1,3-indandione, is the VKA of first choice in France.4 To compare the efficacy of these clinically used VKAs, we examined the IC50 of these anticoagulants for VKOR activity.32 Wild-type VKOR was transiently expressed in the FIXgla-PC/HEK293 DGKO reporter cells, and transfected cells were treated with increasing concentrations of individual VKAs containing 5 µM KO. The efficiency of reporter protein (FIXgla-PC) carboxylation under these conditions was measured using ELISA.34 Our results show that the inhibition curves of warfarin, phenprocoumon, and fluindione are similar (Figure 1B), suggesting a similar inhibition efficiency for VKOR activity. However, acenocoumarol appears to be more efficient in VKOR inactivation, as the inhibition curve shifts toward the lower VKA concentrations. The IC50 of acenocoumarol is approximately sixfold lower than that of other VKAs (Figure 1C; supplemental Table 1, available on the Blood Web site), suggesting that acenocoumarol is the most efficient VKA for anticoagulation. These results are consistent with previous findings showing that warfarin and phenprocoumon are equipotent for VKOR inactivation, but acenocoumarol is more potent than warfarin or phenprocoumon.36
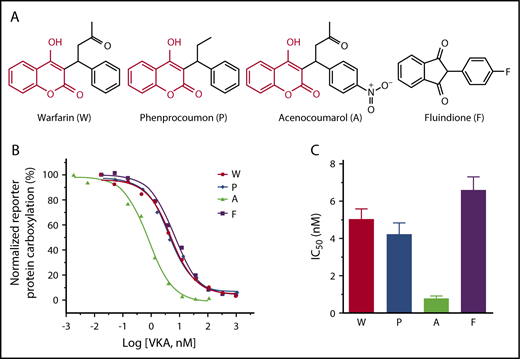
Inhibition of VKOR activity by clinically used VKAs. (A) Chemical structures of the oral anticoagulants warfarin, phenprocoumon, acenocoumarol, and fluindione. The identical chemical structures of 4-hydroxycoumarin in warfarin, phenprocoumon, and acenocoumarol are shown in red. (B) VKA concentration titration against VKOR activity in DGKO FIXgla-PC/HEK293 reporter cells. Wild-type VKOR was transiently expressed in the reporter cells; the transfected cells were incubated with 5 µM KO with increasing concentrations of VKAs. (C) Inhibition efficiency of VKAs on VKOR activity. The IC50 of each VKA was determined from panel B using GraphPad software. Data are presented as mean ± standard deviation (SD).
Inhibition of VKOR activity by clinically used VKAs. (A) Chemical structures of the oral anticoagulants warfarin, phenprocoumon, acenocoumarol, and fluindione. The identical chemical structures of 4-hydroxycoumarin in warfarin, phenprocoumon, and acenocoumarol are shown in red. (B) VKA concentration titration against VKOR activity in DGKO FIXgla-PC/HEK293 reporter cells. Wild-type VKOR was transiently expressed in the reporter cells; the transfected cells were incubated with 5 µM KO with increasing concentrations of VKAs. (C) Inhibition efficiency of VKAs on VKOR activity. The IC50 of each VKA was determined from panel B using GraphPad software. Data are presented as mean ± standard deviation (SD).
Effect of naturally occurring VKOR mutations on warfarin resistance
Twenty-seven naturally occurring VKOR missense mutations have been identified from patients requiring a higher dose of VKAs to reach the desired anticoagulation effect.37 These mutations are referred to as warfarin-resistant mutations. To examine whether these VKOR mutations are the causative factor for the observed warfarin-resistant clinical phenotypes, we determined warfarin resistance for each VKOR mutation by measuring the IC50 of warfarin, as described in the previous section. Our results show that compared with wild-type VKOR, the IC50 values of warfarin toward these VKOR mutants varies from no change (the G71A mutant) to a 98-fold increase (the W59R mutant) (Figure 2; Table 1). Of the 27 clinically identified warfarin-resistant mutations, only 8 mutations (A26P, V29L, A34P, S52L, W59R, W59C, W59L, and L128R) significantly increased warfarin resistance (IC50 increase >20-fold; Figure 2 red bars), whereas 11 mutations only had a minor effect on warfarin resistance (IC50 increase less than threefold; Figure 2 green bars). The other 8 mutations showed a moderate (3.4-fold to 8.5-fold) increase on warfarin resistance (Figure 2 blue bars). As all of the patients bearing these mutations required a high dose of VKAs (warfarin-resistant), our results suggest that not all clinically identified warfarin-resistant VKOR mutations result in a warfarin-resistant VKOR activity, which agrees with previous studies.31,32
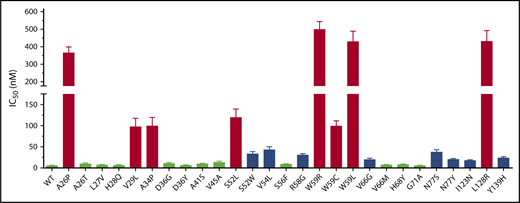
Warfarin-resistance variation among naturally occurring VKOR mutations. Warfarin resistance was evaluated by the IC50. Each individual VKOR mutation was transiently expressed in DGKO FIXgla-PC/HEK293 reporter cells and IC50 was determined, as described in the Figure 1 legend. Green bars, IC50 increase < threefold; blue bars, IC50 increased between 3.4-fold to 8.5-fold; red bars, IC50 increase > 20-fold.
Warfarin-resistance variation among naturally occurring VKOR mutations. Warfarin resistance was evaluated by the IC50. Each individual VKOR mutation was transiently expressed in DGKO FIXgla-PC/HEK293 reporter cells and IC50 was determined, as described in the Figure 1 legend. Green bars, IC50 increase < threefold; blue bars, IC50 increased between 3.4-fold to 8.5-fold; red bars, IC50 increase > 20-fold.
IC50 of clinically used oral anticoagulants for naturally occurring VKOR mutations
| VKOR mutations | IC50 ± standard deviation, nM | |||
|---|---|---|---|---|
| Warfarin | Phenprocoumon | Acenocoumarol | Fluindione | |
| Wild type | 5.1 ± 0.5 | 4.2 ± 0.6 | 0.81 ± 0.12 | 6.6 ± 0.7 |
| A26P | 367 ± 32 | 83.3 ± 6.0 | 7.84 ± 1.13 | 135 ± 42 |
| A26T | 9.8 ± 1.9 | 17.7 ± 2.3 | 1.40 ± 0.16 | 30.2 ± 7.3 |
| L27V | 7.2 ± 0.4 | 8.7 ± 0.8 | 1.25 ± 0.21 | 21.1 ± 1.2 |
| H28Q | 6.6 ± 0.7 | 10.3 ± 1.6 | 0.54 ± 0.06 | 25.9 ± 2.0 |
| V29L | 98 ± 19 | 146 ± 16 | 3.65 ± 0.32 | 95 ± 25 |
| A34P | 99 ± 20 | 128 ± 20 | 1.91 ± 0.23 | 72 ± 17 |
| D36G | 10.5 ± 2.4 | 13.6 ± 1.8 | 0.80 ± 0.09 | 11.5 ± 0.8 |
| D36Y | 6.4 ± 0.8 | 5.1 ± 0.4 | 1.14 ± 0.15 | 9.0 ± 0.2 |
| A41S | 9.5 ± 1.2 | 19.5 ± 1.9 | 0.73 ± 0.07 | 12.8 ± 1.4 |
| V45A | 13.2 ± 2.9 | 17.7 ± 2.2 | 0.91 ± 0.09 | 30.5 ± 1.0 |
| S52L | 119 ± 20 | 87 ± 22 | 2.07 ± 0.14 | 141 ± 16 |
| S52W | 33.5 ± 5.0 | 21.9 ± 4.0 | 1.95 ± 0.21 | 81 ± 13 |
| V54L | 43.2 ± 6.9 | 28.5 ± 1.9 | 6.04 ± 0.79 | 152 ± 19 |
| S56F | 8.4 ± 1.1 | 9.0 ± 2.0 | 1.13 ± 0.07 | 15.0 ± 1.3 |
| R58G | 30.8 ± 3.0 | 14.7 ± 2.8 | 1.16 ± 0.08 | 27.6 ± 3.3 |
| W59R | 500 ± 43 | 835 ± 142 | 11.21 ± 1.47 | 742 ± 76 |
| W59C | 100 ± 12 | 292 ± 23 | 3.81 ± 0.48 | 85.4 ± 5.1 |
| W59L | 431 ± 58 | 398 ± 45 | 6.19 ± 0.79 | 370 ± 58 |
| V66G | 19.9 ± 3.0 | 12.8 ± 2.0 | 1.23 ± 0.16 | 68 ± 11 |
| V66M | 6.8 ± 1.0 | 17.2 ± 1.8 | 1.36 ± 0.14 | 10.2 ± 1.5 |
| H68Y | 8.1 ± 0.9 | 5.5 ± 0.5 | 0.63 ± 0.04 | 12.5 ± 0.6 |
| G71A | 5.2 ± 0.6 | 8.4 ± 1.4 | 1.46 ± 0.26 | 15.2 ± 1.6 |
| N77S | 30.8 ± 5.3 | 53.0 ± 9.6 | 1.45 ± 0.09 | 124 ± 11 |
| N77Y | 19.8 ± 2.5 | 40.5 ± 9.1 | 1.38 ± 0.09 | 23.1 ± 6.4 |
| 123IN | 17.4 ± 2.0 | 46.4 ± 8.2 | 2.60 ± 0.26 | 54.1 ± 8.0 |
| L128R | 431 ± 61 | 592 ± 103 | 10.11 ± 1.62 | 626 ± 127 |
| Y139H | 24.2 ± 2.3 | 60.7 ± 5.8 | 2.27 ± 0.18 | 77.5 ± 6.3 |
| VKOR mutations | IC50 ± standard deviation, nM | |||
|---|---|---|---|---|
| Warfarin | Phenprocoumon | Acenocoumarol | Fluindione | |
| Wild type | 5.1 ± 0.5 | 4.2 ± 0.6 | 0.81 ± 0.12 | 6.6 ± 0.7 |
| A26P | 367 ± 32 | 83.3 ± 6.0 | 7.84 ± 1.13 | 135 ± 42 |
| A26T | 9.8 ± 1.9 | 17.7 ± 2.3 | 1.40 ± 0.16 | 30.2 ± 7.3 |
| L27V | 7.2 ± 0.4 | 8.7 ± 0.8 | 1.25 ± 0.21 | 21.1 ± 1.2 |
| H28Q | 6.6 ± 0.7 | 10.3 ± 1.6 | 0.54 ± 0.06 | 25.9 ± 2.0 |
| V29L | 98 ± 19 | 146 ± 16 | 3.65 ± 0.32 | 95 ± 25 |
| A34P | 99 ± 20 | 128 ± 20 | 1.91 ± 0.23 | 72 ± 17 |
| D36G | 10.5 ± 2.4 | 13.6 ± 1.8 | 0.80 ± 0.09 | 11.5 ± 0.8 |
| D36Y | 6.4 ± 0.8 | 5.1 ± 0.4 | 1.14 ± 0.15 | 9.0 ± 0.2 |
| A41S | 9.5 ± 1.2 | 19.5 ± 1.9 | 0.73 ± 0.07 | 12.8 ± 1.4 |
| V45A | 13.2 ± 2.9 | 17.7 ± 2.2 | 0.91 ± 0.09 | 30.5 ± 1.0 |
| S52L | 119 ± 20 | 87 ± 22 | 2.07 ± 0.14 | 141 ± 16 |
| S52W | 33.5 ± 5.0 | 21.9 ± 4.0 | 1.95 ± 0.21 | 81 ± 13 |
| V54L | 43.2 ± 6.9 | 28.5 ± 1.9 | 6.04 ± 0.79 | 152 ± 19 |
| S56F | 8.4 ± 1.1 | 9.0 ± 2.0 | 1.13 ± 0.07 | 15.0 ± 1.3 |
| R58G | 30.8 ± 3.0 | 14.7 ± 2.8 | 1.16 ± 0.08 | 27.6 ± 3.3 |
| W59R | 500 ± 43 | 835 ± 142 | 11.21 ± 1.47 | 742 ± 76 |
| W59C | 100 ± 12 | 292 ± 23 | 3.81 ± 0.48 | 85.4 ± 5.1 |
| W59L | 431 ± 58 | 398 ± 45 | 6.19 ± 0.79 | 370 ± 58 |
| V66G | 19.9 ± 3.0 | 12.8 ± 2.0 | 1.23 ± 0.16 | 68 ± 11 |
| V66M | 6.8 ± 1.0 | 17.2 ± 1.8 | 1.36 ± 0.14 | 10.2 ± 1.5 |
| H68Y | 8.1 ± 0.9 | 5.5 ± 0.5 | 0.63 ± 0.04 | 12.5 ± 0.6 |
| G71A | 5.2 ± 0.6 | 8.4 ± 1.4 | 1.46 ± 0.26 | 15.2 ± 1.6 |
| N77S | 30.8 ± 5.3 | 53.0 ± 9.6 | 1.45 ± 0.09 | 124 ± 11 |
| N77Y | 19.8 ± 2.5 | 40.5 ± 9.1 | 1.38 ± 0.09 | 23.1 ± 6.4 |
| 123IN | 17.4 ± 2.0 | 46.4 ± 8.2 | 2.60 ± 0.26 | 54.1 ± 8.0 |
| L128R | 431 ± 61 | 592 ± 103 | 10.11 ± 1.62 | 626 ± 127 |
| Y139H | 24.2 ± 2.3 | 60.7 ± 5.8 | 2.27 ± 0.18 | 77.5 ± 6.3 |
Resistance of other VKAs toward naturally occurring VKOR mutations
To examine the effect of naturally occurring VKOR mutations on the resistance of other clinically used VKAs, we extended this study to determine the IC50 of acenocoumarol, phenprocoumon, and fluindione for each of the 27 VKOR mutations. Our results (Table 1; supplemental Figure 1) show that, in general, the relative resistance of VKOR mutations to these 3 VKAs is similar to that of warfarin. The most warfarin-resistant mutations (W59R, W59L, and L128R) are also highly resistant to inhibition by these 3 VKAs. W59R is the most resistant mutation of all 4 VKAs. On the other hand, the relative resistance of some of the mutations to these VKAs is significantly different. For example, the A26P mutant shows a dramatically increased relative resistance to warfarin and acenocoumarol, but has only a slightly increased relative resistance to phenprocoumon and fluindione. The V54L mutant is highly resistant to acenocoumarol, but is significantly less resistant to all other VKAs. The resistance variations of different VKAs toward these VKOR mutations may relate to their chemical structure differences and/or to their possibly different inhibition mechanisms, as discussed in “Warfarin and fluindione appear to have different mechanisms for VKOR inhibition.”
To better understand the resistance variations of different VKAs among the VKOR mutations, we compared the relative resistance of these VKAs by normalizing the IC50 of each VKA toward wild-type VKOR as one. Results in Figure 3 show that the most resistant VKOR mutation, W59R, shows a 199-fold increase in resistance for phenprocoumon, an ∼100-fold increase in resistance for warfarin and fluindione, and only a 14-fold increase in resistance for acenocoumarol. Acenocoumarol had the least resistance variation suggesting that in order to reach a certain anticoagulation effect, acenocoumarol should have the smallest dosage variation among patients carrying different VKOR mutations.
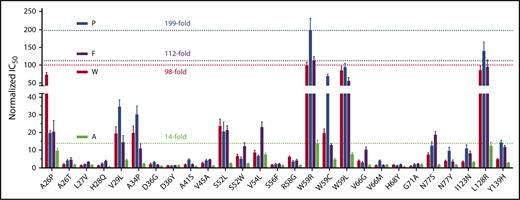
Comparison of the relative resistance variations of all 4 VKAs among 27 naturally occurring VKOR mutations. The IC50 of phenprocoumon (P), acenocoumarol (A), and fluindione (F) for all 27 VKOR mutations was determined, as described in the Figure 1 legend. The IC50 of each VKA for wild-type VKOR was normalized to 1. The most resistant level among VKOR mutations of each VKA is indicated by a dotted line (also indicated by the fold increase) in the same color as their respective bar graphs.
Comparison of the relative resistance variations of all 4 VKAs among 27 naturally occurring VKOR mutations. The IC50 of phenprocoumon (P), acenocoumarol (A), and fluindione (F) for all 27 VKOR mutations was determined, as described in the Figure 1 legend. The IC50 of each VKA for wild-type VKOR was normalized to 1. The most resistant level among VKOR mutations of each VKA is indicated by a dotted line (also indicated by the fold increase) in the same color as their respective bar graphs.
Effect of naturally occurring VKOR mutations on VKOR activity
It has been reported that several naturally occurring VKOR mutations abolished VKOR activity when measured by the conventional in vitro activity assay.29,31 As patients bearing these mutations do not have bleeding phenotypes under normal conditions,38 we suspect that these mutations should not have a dramatic effect on VKOR activity under physiological conditions. To test this hypothesis, we determined the VKOR activity of the naturally occurring VKOR mutations in a cellular environment using 5 µM KO.32 Results in Figure 4A show that all of these VKOR mutations appear to be fully functional, which is consistent with the patient’s nonbleeding phenotypes.
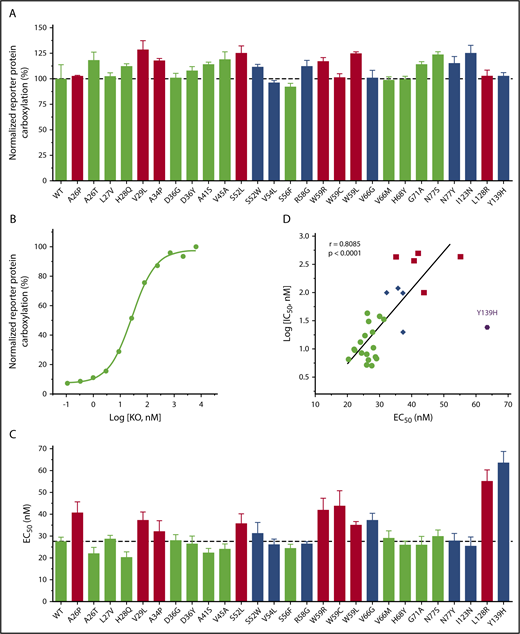
Effect of naturally occurring VKOR mutations on VKOR activity and KO binding. (A) Cell-based VKOR activity of all naturally occurring VKOR mutations. Each VKOR mutation was transiently expressed in DGKO FIXgla-PC/HEK293 reporter cells; the transfected cells were cultured with 5 µM KO for activity assay. Bar color codes are the same as in Figure 2. Wild-type VKOR activity is indicated by the dotted line. (B) KO concentration titration of wild-type VKOR transiently expressed in DGKO FIXgla-PC/HEK293 reporter cells. The EC50 of KO was determined by GraphPad software. (C) The EC50 of KO for all naturally occurring VKOR mutations. Data are presented as mean ± SD. Bar color codes are the same as Figure 2. The EC50 of the wild-type VKOR is indicated by the dotted line. (D) Correlation between IC50 of warfarin and EC50 of KO for naturally occurring VKOR mutations. Data for the Y139H mutant were excluded for the correlation calculation.
Effect of naturally occurring VKOR mutations on VKOR activity and KO binding. (A) Cell-based VKOR activity of all naturally occurring VKOR mutations. Each VKOR mutation was transiently expressed in DGKO FIXgla-PC/HEK293 reporter cells; the transfected cells were cultured with 5 µM KO for activity assay. Bar color codes are the same as in Figure 2. Wild-type VKOR activity is indicated by the dotted line. (B) KO concentration titration of wild-type VKOR transiently expressed in DGKO FIXgla-PC/HEK293 reporter cells. The EC50 of KO was determined by GraphPad software. (C) The EC50 of KO for all naturally occurring VKOR mutations. Data are presented as mean ± SD. Bar color codes are the same as Figure 2. The EC50 of the wild-type VKOR is indicated by the dotted line. (D) Correlation between IC50 of warfarin and EC50 of KO for naturally occurring VKOR mutations. Data for the Y139H mutant were excluded for the correlation calculation.
To explore the details of how these mutations might affect VKOR’s substrate (KO) binding, we determined the EC50 (or apparent Km) of KO for all 27 VKOR mutations. EC50 was determined by the KO titration of DGKO reporter cells transiently expressing the individual VKOR mutant. Figure 4B shows an example of KO titration for wild-type VKOR. Results in Figure 4C and supplemental Table 2 show that these VKOR mutations have only a minor effect on the apparent Km of KO (KO binding) (EC50 increase less than threefold), although some of these mutations dramatically decreased the warfarin binding (IC50 increased 20-fold to 98-fold) (Figure 3). On the other hand, VKOR mutations with a minor effect on warfarin binding (Figure 2 green bars), have similar EC50 values as wild-type VKOR (Figure 4C green bars). This suggests that these clinically identified warfarin-resistant VKOR mutations do not affect either warfarin or KO binding. Meanwhile, VKOR mutations that dramatically decreased warfarin binding (Figure 2 red bars) also showed increased EC50 values (Figure 4C red bars), suggesting that these mutations affect both warfarin and KO bindings. In general, there is a correlation between the effect of VKOR mutations on warfarin and KO binding (Figure 4D), although the effect on warfarin binding (a maximal IC50 increase of 98-fold) is significantly greater than KO binding (a maximal EC50 increase of 2.3-fold). Interestingly, the Y139H mutant results in the most significant effect on KO binding of all the mutations tested (Figure 4C), but has only a minor effect on warfarin binding (IC50 increased 4.7-fold vs the maximal of 98-fold) (Figure 2; supplemental Table 1). The significant effect of the Y139H mutation on KO binding could be related to the close location of residue Y139 to VKOR’s C132XXC135 active site where KO is reduced. Overall, these results suggest that naturally occurring VKOR mutations respond differently to warfarin and KO binding.
Warfarin and fluindione appear to have different mechanisms for VKOR inhibition
Although warfarin and fluindione are structurally different (Figure 1A), these 2 VKAs have similar efficiencies in VKOR inhibition (Figure 1B-C) and similar resistance variations among naturally occurring VKOR mutations (Figure 3). To explore the possible differences of the inhibition mechanisms between these 2 VKAs, we determined the apparent Michaelis-Menten kinetic parameters associated with KO as a function of different concentrations of warfarin or fluindione using our cell-based assay.34 Our results show that fluindione, at different concentrations, significantly affects the apparent Km of KO, but not the apparent maximum velocity (Vmax) of KO reduction (Figure 5A; supplemental Table 3). This result suggests that fluindione is likely to be a competitive inhibitor of VKOR,39 which is consistent with the results of plotting the experimental data to the Lineweaver-Burk plot (Figure 5B), showing that the plotted lines of the control (without inhibitor) and of the different concentrations of the inhibitor intersect at the y-axis. The competitive inhibition of VKOR by fluindione is more apparent when the results are visualized by the Cornish-Bowden plot, which shows that the plotted lines for different substrate concentrations are nearly parallel to each other (Figure 5C).40 Furthermore, when all data were used to fit different inhibition models using nonlinear regression by GraphPad software,41 competitive inhibition is the best fit (supplemental Figure 2; supplemental Table 4). These results together suggest that the inhibition of fluindione to VKOR is a competitive inhibition.
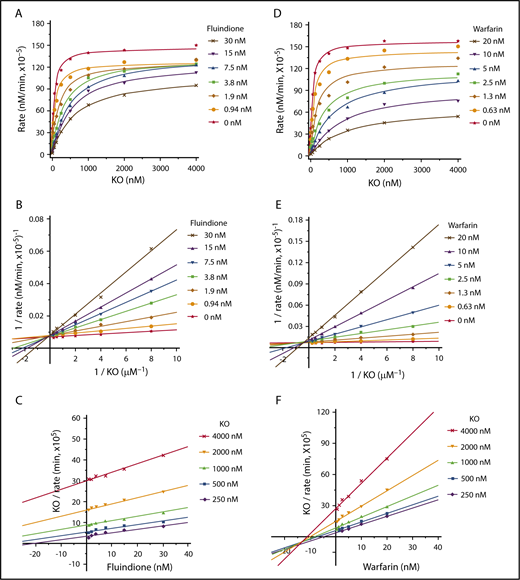
Cell-based kinetics study of the inhibition of VKOR by warfarin and fluindione. (A) Michaelis-Menten plot of VKOR activity as a function of KO concentration in the absence and presence of fluindione. FIXgla-PC/HEK293 reporter cells were cultured with increasing concentrations of KO containing 0, 0.94, 1.88, 3.75, 7.5, 15, and 30 nM fluindione for the activity assay. (B) Lineweaver-Burk plot of the experimental data of panel A. (C) Cornish-Bowden plot of the experimental data of panel A. (D) Michaelis-Menten plot of VKOR activity as a function of KO concentrations in the absence and presence of warfarin. FIXgla-PC/HEK293 reporter cells were cultured with increasing concentrations of KO containing 0, 0.62, 1.25, 2.5, 5, 10, and 20 nM warfarin for the activity assay. (E) Lineweaver-Burk plot of the experimental data of panel D. (F) Cornish-Bowden plot of the experimental data of panel D.
Cell-based kinetics study of the inhibition of VKOR by warfarin and fluindione. (A) Michaelis-Menten plot of VKOR activity as a function of KO concentration in the absence and presence of fluindione. FIXgla-PC/HEK293 reporter cells were cultured with increasing concentrations of KO containing 0, 0.94, 1.88, 3.75, 7.5, 15, and 30 nM fluindione for the activity assay. (B) Lineweaver-Burk plot of the experimental data of panel A. (C) Cornish-Bowden plot of the experimental data of panel A. (D) Michaelis-Menten plot of VKOR activity as a function of KO concentrations in the absence and presence of warfarin. FIXgla-PC/HEK293 reporter cells were cultured with increasing concentrations of KO containing 0, 0.62, 1.25, 2.5, 5, 10, and 20 nM warfarin for the activity assay. (E) Lineweaver-Burk plot of the experimental data of panel D. (F) Cornish-Bowden plot of the experimental data of panel D.
However, when warfarin was used as an inhibitor, both the apparent Km and Vmax were affected (Figure 5D; supplemental Table 5). Increasing warfarin concentrations in the reaction decreased the apparent Vmax and increased the apparent Km, suggesting that warfarin, unlike fluindione, is not a competitive inhibitor of VKOR. A Lineweaver-Burk plot of the experimental data shows that plotted lines intersect to the left of the y-axis and above the x-axis (Figure 5E), suggesting mixed inhibition. The mixed inhibition of VKOR by warfarin is also supported by the Cornish-Bowden plot40 showing that the plotted lines intersect to the left of the y-axis and below the x-axis (Figure 5F). When data for all concentrations were fit simultaneously to different inhibition models using nonlinear regression by GraphPad software, the best fit is mixed inhibition39,42 (supplemental Figure 3; supplemental Table 6). Together, these results suggest that warfarin and fluindione have different mechanisms of VKOR inhibition.
Discussion
Our initial goal was to evaluate the efficacy of clinically used oral anticoagulants and to elucidate the dosage variations of VKAs among patients with genetic variations in VKOR, the target enzyme of VKAs. As functional studies of the naturally occurring VKOR mutations using conventional in vitro activity assays gave controversial results, we explored these questions using our recently established cell-based assays.32,34
We first compared how VKOR activity was inhibited by warfarin, acenocoumarol, phenprocoumon, and fluindione. Our results showed that the concentrations required for the half-maximal inhibition of VKOR activity follows the order of: acenocoumarol < phenprocoumon < warfarin < fluindione (Figure 1C). Considering the significantly different half-life of these VKAs and other clinical factors that affect VKA dosages,1 our results are consistent, in general, with the clinical mean VKA dose used for anticoagulation control.43-45 In real-world anticoagulation therapy, all 4 VKAs were used in the same patient bearing a heterozygous mutation of L128R in VKOR.28,46 The daily dosage of these VKAs to achieve the desired INR (1.0-1.5) correlates well with the relative efficiency (IC50) of the VKAs obtained from our cell-based assay (Figure 6A). For example, to achieve an INR of ∼1.2, fluindione has the highest daily dose of 80 mg,28,46 which is consistent with our cell-based assay results showing that fluindione has the largest IC50 (6.6 nM) among the 4 VKAs. Additionally, the effective daily dose of fluindione is eightfold higher than that of acenocoumarol (10 mg per day), which again agrees well with the 8.1-fold higher IC50 of fluindione (6.6 nM) than acenocoumarol (0.81 nM). These results suggest that our cell-based assay is a good model for studying VKAs targeting VKOR under physiological conditions.
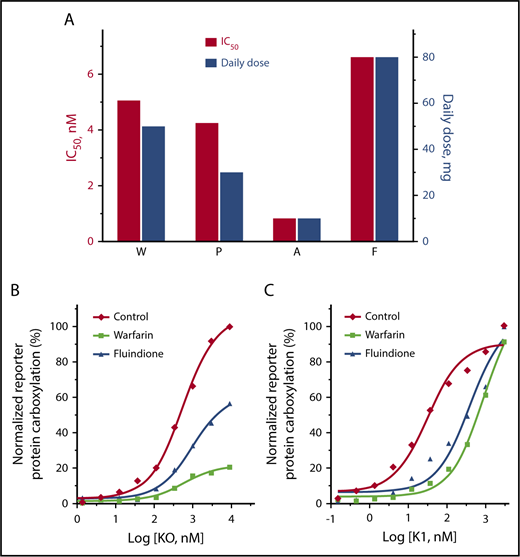
Comparison of VKA anticoagulation efficacy and the reversal anticoagulation effect of KO and vitamin K. (A) Comparison of the cell-based inhibition efficiency (IC50) of the 4 VKAs on VKOR activity and the effective clinical dosages of these VKAs when used in the same patient for anticoagulation control. Clinical daily dosages of the VKAs were obtained from Johnson et al26 and Undas et al.43 (B-C) Cell-based functional study of reversing the anticoagulation effect of warfarin and fluindione by KO (B) or vitamin K (C). FIXgla-PC/HEK293 reporter cells were seeded in a multiwell cell culture plate with complete medium alone (Control) or complete medium containing 100 nM fluindione (F) or 100 nM warfarin (W). Twenty-four hours later, cell culture media were replaced with the same medium containing increasing concentrations of KO (B) or vitamin K (C). These cells were cultured another 24 hours for activity assay.
Comparison of VKA anticoagulation efficacy and the reversal anticoagulation effect of KO and vitamin K. (A) Comparison of the cell-based inhibition efficiency (IC50) of the 4 VKAs on VKOR activity and the effective clinical dosages of these VKAs when used in the same patient for anticoagulation control. Clinical daily dosages of the VKAs were obtained from Johnson et al26 and Undas et al.43 (B-C) Cell-based functional study of reversing the anticoagulation effect of warfarin and fluindione by KO (B) or vitamin K (C). FIXgla-PC/HEK293 reporter cells were seeded in a multiwell cell culture plate with complete medium alone (Control) or complete medium containing 100 nM fluindione (F) or 100 nM warfarin (W). Twenty-four hours later, cell culture media were replaced with the same medium containing increasing concentrations of KO (B) or vitamin K (C). These cells were cultured another 24 hours for activity assay.
Next, we examined the effect of clinically identified VKA-resistant VKOR mutations on the resistance of the 4 oral anticoagulants. Compared with wild-type VKOR, the W59R, W59L, and L128R mutants showed a significantly increased resistance to all 4 VKAs (Figure 3). This result is consistent with clinical observations that patients bearing these mutations require significantly higher doses of VKAs to achieve the desired anticoagulation effect37 ; some patients even exhibit an apparently “complete” resistance to VKA therapy. The relative resistance of phenprocoumon among the VKOR mutations varies from 1.2-fold (D36Y) to 199-fold (W59R), whereas the variation of the relative resistance of acenocoumarol among the VKOR mutations is only 14-fold. Together with the results of the inhibition efficiency of these oral anticoagulants (Figure 1), these results suggest that acenocoumarol is the most efficient VKA with the least resistance variation among VKOR mutations. These cell-based assay results are consistent with clinical observations showing that acenocoumarol is an effective and safe anticoagulant that offers an advantage over warfarin in terms of better stability of the anticoagulant effect.47 Therefore, results from this study provide new biochemical insights into dosage management of commonly used VKAs when the patient’s genetic information of VKOR is available.
It is worth noting that of the 27 clinically identified VKA-resistant VKOR mutations, 11 mutations only show a negligible difference in VKA sensitivity as compared with that of the wild-type enzyme. Additionally, VKA sensitivity of some of the VKOR mutations do not correlate with the clinical VKA dosages of patients carrying these mutations. For example, a patient with a V45A mutation was completely resistant to warfarin therapy,29,37 whereas the warfarin sensitivity of the V45A mutant is only 2.6-fold lower than that of wild-type VKOR. Additionally, patients bearing a D36Y mutation required larger doses of warfarin than patients carrying an A26P mutation in order to achieve a stable anticoagulation effect.28,37,48 However, warfarin sensitivity of the A26P mutant decreased over 72-fold compared with the wild-type enzyme, whereas warfarin sensitivity of the D36Y mutant only showed a 1.3-fold decrease. These results suggest that not all clinically identified VKA-resistant VKOR mutations cause VKA-resistant VKOR activity. This is consistent with previous observations using both in vitro– and cell-based VKOR activity assays.31,32 VKA-resistant phenotypes have been reported to be associated with other clinical factors, including patient’s physical conditions, their diet, drug interactions, and, importantly, the polymorphism of the VKA metabolism enzymes (such as CYP2C9).49,50 Therefore, factors other than genetic variations in VKOR should be considered when interpreting VKA-resistant clinical phenotypes, especially for patients carrying VKOR mutations that show a non-VKA-resistant VKOR activity.
Of the 4 clinically used VKAs, warfarin, acenocoumarol, and phenprocoumon are 4-hydroxycoumarin derivatives, whereas fluindione is a derivative of 1,3-indandione (Figure 1A). The mechanisms of how these VKAs inactivate their target enzyme VKOR are not well defined. The mechanism of warfarin’s inhibition of VKOR has been extensively studied using conventional in vitro VKOR activity assay. However, both irreversible51,52 and reversible36 inhibition mechanisms have been proposed. As a reversible inhibitor, most of the studies support warfarin as a noncompetitive inhibitor.31,53,54 It should be noted that these studies were performed using VKOR-containing microsomes, and because the physiological reductant of VKOR is unknown, dithiothreitol was used to regenerate the active site of VKOR for their in vitro activity assay.
Using a cell-based enzyme kinetics study and a conventional in vitro VKOR activity assay, we recently showed that warfarin’s inhibition of VKOR is reversible.55 We extended these observations to include structurally different oral anticoagulants in order to better understand the mechanism of VKOR inhibition. Our results suggest that warfarin appears to inhibit VKOR by a mixed type inhibition (Figure 5). This could mean that warfarin’s inhibition of VKOR is a combination of noncompetitive and competitive inhibitions.39,56 However, fluindione, a structurally different VKA, appears to exhibit a competitive inhibition mode for VKOR inactivation. The mechanistically different inhibitions between warfarin and fluindione imply that the inactivation of VKOR by fluindione could be efficiently reversed by high substrate (KO) concentrations, but not the inhibition by warfarin (Figure 6B). However, in anticoagulation therapy, patients that are overdosed with warfarin, a mixed inhibitor, can be rescued by administering large doses of vitamin K.57-59 It should be noted that the mechanism of VKA inhibition of VKOR observed in this study only applies to the reduction of KO to vitamin K, but not the reduction of vitamin K to KH2, as there is a VKA-resistant vitamin K reductase in HEK293 cells.34,60 The antidotal effect of vitamin K is mainly due to the existence of the unknown VKA-resistant vitamin K reductase that directly converts vitamin K to KH2.34,61 As shown in Figure 6C, the inhibition of vitamin K–dependent carboxylation by both warfarin and fluindione can be fully reversed by higher concentrations of vitamin K, although a lower vitamin K concentration is required for recovering fluindione inhibition than for recovering warfarin inhibition. These results suggest that fluindione is more responsive than warfarin with respect to reversal of the anticoagulant effect by vitamin K.
In summary, we have examined the efficacy of the clinically used VKAs under physiological conditions using our recently established cell-based VKOR activity assay. We also compared the effect of naturally occurring VKOR mutations on the anticoagulation effect of each of the VKAs. Our results suggest that acenocoumarol is the most efficient VKA with the least variations of resistance among the VKOR mutations. Warfarin and fluindione have similar anticoagulation efficiencies, but they appear to have different mechanisms for VKOR inhibition. It should be noted that results from this study are solely based on the inactivation of VKOR, or its naturally occurring mutants, by clinically used VKAs in a cellular environment. The efficacy of these VKAs on anticoagulation therapy can be affected by other factors as well, as previously discussed.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Lee Pedersen for the helpful discussion and for proofreading the manuscript.
This work was supported by grant HL131690 from the National Institutes of Health, National Heart, Lung, and Blood Institute (J.-K.T. and D.W.S.).
Authorship
Contribution: X.C. performed all of the cell-based functional studies and analyzed the data; D.-Y.J. created the reporter cell lines and made all of the VKOR mutation constructs; D.W.S. oversaw the whole project; and J.-K.T. designed the study, analyzed the data, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Jian-Ke Tie, Department of Biology, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599; e-mail: jktie@email.unc.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal