

Abstract
Mantle cell lymphoma (MCL) is a rare subtype of non-Hodgkin lymphoma that is most commonly treated with combination chemo-immunotherapy at diagnosis because of the poor prognosis. More indolent presentations have been described including patients who can defer initial therapy without adverse impact on survival. The 2016 World Health Organization updated classification describes 2 major subtypes, classical and leukemic nonnodal MCL, each with unique molecular features and clinical presentations. Although there is no standard of care for MCL, aggressive chemo-immunotherapy regimens containing rituximab and cytarabine, followed by consolidation with autologous stem cell transplantation and maintenance rituximab, are the most used approach in young fit patients, and chemo-immunotherapy, followed by rituximab maintenance, is most commonly used in older patients. Despite the improvement in response durations with currently available therapies, patients will inevitably relapse. A number of targeted therapies are approved in the relapsed setting and are now under evaluation in combination with standard frontline therapy. Although the approval of ibrutinib changed the landscape of therapy for relapsed MCL, prognosis remains poor after progression on ibrutinib supporting the development of ibrutinib combinations to prolong response duration as well as the development of other novel agents for ibrutinib refractory disease. With ibrutinib being incorporated into initial therapy regimens, new options will be needed at relapse. Prognostic markers, such as minimal residual disease, have been shown to correlate independently with outcomes along with predicting relapse, with the potential to guide therapeutic decisions. The future treatment of MCL therapy will need to incorporate therapy based on risk-stratification and nonchemotherapeutic approaches.
Introduction
Mantle cell lymphoma (MCL) is a rare incurable B-cell lymphoma defined by the translocation (11;14)(q13;q32) and resulting in constitutive overexpression of cyclin D1.1 Historically recognized as an aggressive lymphoma requiring treatment at diagnosis, MCL is a heterogeneous disease with variable presentations, clinical and biologic risk factors, and treatment approaches (Figure 1). Despite an improved understanding of biology and the development of effective therapeutic strategies resulting in improved survival, patients continue to have a poor prognosis overall. A better understanding of how to incorporate novel therapies and utilization of risk-adapted treatment approaches will be critical to further improving outcomes in this disease.
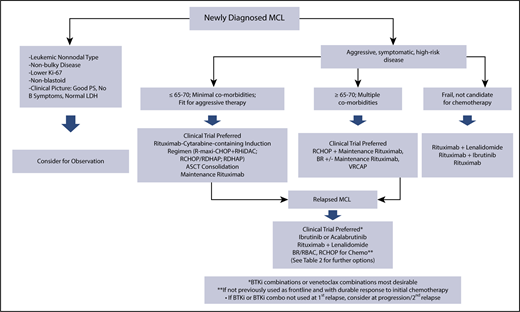
MCL treatment algorithm. ASCT, autologous stem cell transplantation; BR, bendamustine and rituximab; BTKi, Bruton’s tyrosine kinase inhibitor; LDH, lactate dehydrogenase; PS, performance status; RBAC, rituximab, bendamustine, and cytarabine; RCHOP, rituximab, cyclophosphamide, vincristine, doxorubicin, and prednisone; RDHAP, rituximab, dexamethasone, cytarabine, and cisplatin; RHiDAC, rituximab, high-dose cytarabine; R-maxi-CHOP, dose intensified cyclophosphamide, doxorubicin, vincristine, prednisone; VRCAP, cyclophosphamide, doxorubicin, bortezomib, and prednisone.
MCL treatment algorithm. ASCT, autologous stem cell transplantation; BR, bendamustine and rituximab; BTKi, Bruton’s tyrosine kinase inhibitor; LDH, lactate dehydrogenase; PS, performance status; RBAC, rituximab, bendamustine, and cytarabine; RCHOP, rituximab, cyclophosphamide, vincristine, doxorubicin, and prednisone; RDHAP, rituximab, dexamethasone, cytarabine, and cisplatin; RHiDAC, rituximab, high-dose cytarabine; R-maxi-CHOP, dose intensified cyclophosphamide, doxorubicin, vincristine, prednisone; VRCAP, cyclophosphamide, doxorubicin, bortezomib, and prednisone.
Disease classification
The 2016 World Health Organization updated classification recognized 2 molecular pathways of MCL development accounting for different clinical presentations.2 Classical MCL is composed of mature B cells that do not enter the germinal center, have no or minimal mutations in IGHV, and express transcription factor SOX11. Patients present clinically with involvement of lymph nodes and extranodal sites. These tumors are prone to acquisition of additional abnormalities in cell cycle dysregulation, DNA damage response, or cell survival pathways, leading to more aggressive disease behavior, including blastoid and pleomorphic histology. Less commonly, leukemic nonnodal MCL develops through the germinal center with IGHV somatic hypermutation and lack of, or minimal, SOX11 expression. Patients present with involvement of peripheral blood, bone marrow, and spleen. Leukemic nonnodal MCL behaves in a more indolent fashion, with genetic stability over time; however, secondary genetic abnormalities, such as TP53 mutations, result in more aggressive disease associated with poor outcome.
Prognostic markers
A number of clinical and biologic features are prognostic in MCL; the most important routinely available independent factors are the MCL International Prognostic Index (MIPI)3 and tumor cell proliferation rate by Ki-67.4,5 Hoster et al evaluated the prognostic impact of growth pattern and cytology, along with Ki-67 and MIPI, in patients treated in the European MCL Younger and Elderly Trials to define MIPI-c.6 Blastoid cytology was associated with inferior progression-free survival (PFS) and overall survival (OS), with only OS independent of MIPI. They found growth pattern to have minimal prognostic relevance, not independent of other factors. Combining independent risk factors of MIPI and Ki-67, they defined 4 risk groups with 5-year OS ranging from 17% to 85%. This was applicable across patients treated with or without autologous stem cell transplant (ASCT). Although prognostic with standard therapy, MIPI-c has not been prospectively studied to drive therapeutic decisions, and its relevance to novel therapies is unknown.
A number of genetic abnormalities are associated with inferior outcomes with standard therapy. In the European MCL Younger trial, deletions in TP53 and CDKN2A were associated with shorter OS (median, 1.8 vs 7 years) when both deletions were present compared with neither deletion being present.7 In the Nordic MCL2 and MCL3 trials, these deletions, along with TP53 and NOTCH1 mutations, were associated with inferior outcomes; however, multivariate analysis confirmed that only TP53 mutations maintained independent prognostic impact on OS (median, 1.8 vs 12.7 years).8 TP53-mutated cases were associated with blastoid morphology, high Ki-67, high-risk MIPI, and MIPI-c. Patients with TP53 mutations had inferior outcomes with respect to response after both induction and ASCT and inferior PFS compared with TP53-unmutated patients defining a very high-risk population.
The Lymphoma/Leukemia Molecular Profiling Project recently reported on an MCL-proliferation signature based on microarray gene-expression profiling using NanoString technology.9 The assay used pretreatment formalin-fixed paraffin-embedded biopsies from a cohort of 110 MCL patients uniformly treated with rituximab, cyclophosphamide, vincristine, doxorubicin, and prednisone (RCHOP). Patients were identified to be high risk (26%), standard risk (29%), or low risk (45%) based on proliferation signature score, which was predictive of OS (1.1, 2.6, and 8.6 years, respectively) and was independent of MIPI. This proliferation signature requires validation in larger uniformly treated cohorts, but it has potential for prospective study in guiding treatment options.
Despite several clinical and biologic features predicting high-risk disease behavior and inferior outcomes to available therapies, these prognostic markers are not currently used to guide therapy. These features identify patients who should be prioritized for novel frontline therapeutic strategies and investigational combinations.
Current and emerging treatment strategies
Observation
The majority of patients are treated at diagnosis because of disease-related symptoms, involved sites, or rapidity of progression. It is now recognized that a subset of patients can be safely observed, some for prolonged periods. Martin et al first published outcomes on patients deferring initial therapy for >3 months after diagnosis.10 Median time to treatment in these patients was 12 months (range, 4-128), with survival superior to patients treated within 3 months of diagnosis (OS, not reached vs 64 months). More recently, the British Columbia Cancer Agency published on 440 newly diagnosed patients, including 75 (17%) patients observed for ≥3 months, with median time to treatment of 35 months (range, 5-79).11 Median OS was significantly longer in the observation group (72 vs 52.5 months, P = .041) and was not compromised by deferring therapy. Factors associated with deferred treatment included good performance status, no B symptoms, normal lactate dehydrogenase, nonbulky disease, nonblastoid morphology, and lower Ki-67. Other retrospective series have confirmed that deferred therapy in select patients does not impact outcomes.12 Although the majority of patients progress to require therapy, deferring treatment is appropriate in ∼20% of patients with indolent disease behavior and biology, including leukemic nonnodal presentation with indolent disease.2 Clinical trials evaluating novel therapies are needed to address whether targeted treatment approaches are as effective as intensive therapy or could be used to delay treatment with intensive therapy, recognizing that less intensive therapies may negatively impact disease behavior and biology by selecting more resistant clones.
Frontline therapy
There is no standard frontline therapy for MCL. Several regimens prolong response durations (Table 1), but none is curative. The current treatment approach is based on patient age and fitness, with fit patients receiving intensive combination therapies incorporating rituximab and cytarabine, with or without consolidation ASCT, whereas older or unfit patients are treated with combination chemo-immunotherapy, with or without rituximab maintenance (MR). Emerging strategies seek to incorporate newer therapies like ibrutinib into frontline treatment and use prognostic markers, such as minimal residual disease (MRD), to guide therapy decisions.
Selected frontline treatment regimens
| Induction | Consolidation | N | ORR (CR), % | FFS/PFS | Median OS | Toxicities of interest or significance | TRM, % |
|---|---|---|---|---|---|---|---|
| Transplant eligible | |||||||
| Phase 2 | |||||||
| R-HyperCVAD13,17 | NA | 97 | 97 (87) | 5-y 49% FFS | 5-y 52% | 4 AML/MDS | 8 |
| 15-y 22% FFS | 15-y 33% | 5 deaths from acute toxicity | |||||
| R-HyperCVAD15 | NA | 49 | 86 (58) | 5-y 49% PFS | 5-y 63% | 39% did not complete treatment Hematologic and infectious 2 MDS 1 death from acute toxicity | 2 |
| R-HyperCVAD16 | NA | 60 | 83 (72) | 5-y 61% PFS | 5-y 73% | 63% did not complete treatment Hematologic and infectious 3 deaths from acute toxicity | 6.5 |
| R-maxi-CHOP+RHiDAC20,21 | ASCT | 160 | 94 (54) | 6-y 66% PFS | 6-y 70% | 5 did not proceed to ASCT due to toxicity 1 MDS | 5 |
| ASCT | 60 | 95 (57) | 7 y EFS | 5-y 75% | 3 did not proceed to ASCT due to toxicity | 1.7 | |
| BR/RHiDAC29 | ASCT | 23 | 96 (96) | 1-y 96 | 1-y 100% | 0 | |
| Phase 3 | |||||||
| RCHOP vs RCHOP/RDHAP25 | ASCT | 455 | 98 vs 97 (63 vs 61) | 3.8-y PFS vs 7.3-y PFS | 6.8 y vs not reached | Hematologic and infectious | 4 |
| RDHAP28 | ASCT vs ASCT + MR | 299 | 4-y PFS 64% vs 83% | 4-y OS 89% vs 80% | 2 failed stem cell collection 38% received other platinum drug with first cycle | ||
| Transplant ineligible | |||||||
| Phase 3 | |||||||
| RCHOP vs RFC38 | NA | 455 | 86 vs 78 | TTF 28 mo vs 26 mo | 4-y 62% vs 47% | Primary hematologic | |
| 34 vs 40 | |||||||
| RCHOP vs BR40,41 | NA | 94 | 91 vs 93 | 22.1 vs 35.4 mo (median) | Median not reached | ||
| 30 vs 40 | Median not reached | ||||||
| RCHOP vs VRCAP43 | NA | 487 | 89 vs 92 (42 vs 53) | 14.4 vs 24.7 mo | 4-y 54% vs 64% | Primary hematologic | |
| Phase 2 | |||||||
| RBAC 50044 | NA | 57 | 91 | 2-y PFS 81% | Median not reached | Primary hematologic | |
| Nonchemotherapeutic | |||||||
| Rituximab + lenalidomide46,47 | LR | 38 | 92 | 4-y PFS 70% | 4-y 83% | Primary hematologic | |
| 64 | Rash; tumor flare | ||||||
| 7 secondary malignancies |
| Induction | Consolidation | N | ORR (CR), % | FFS/PFS | Median OS | Toxicities of interest or significance | TRM, % |
|---|---|---|---|---|---|---|---|
| Transplant eligible | |||||||
| Phase 2 | |||||||
| R-HyperCVAD13,17 | NA | 97 | 97 (87) | 5-y 49% FFS | 5-y 52% | 4 AML/MDS | 8 |
| 15-y 22% FFS | 15-y 33% | 5 deaths from acute toxicity | |||||
| R-HyperCVAD15 | NA | 49 | 86 (58) | 5-y 49% PFS | 5-y 63% | 39% did not complete treatment Hematologic and infectious 2 MDS 1 death from acute toxicity | 2 |
| R-HyperCVAD16 | NA | 60 | 83 (72) | 5-y 61% PFS | 5-y 73% | 63% did not complete treatment Hematologic and infectious 3 deaths from acute toxicity | 6.5 |
| R-maxi-CHOP+RHiDAC20,21 | ASCT | 160 | 94 (54) | 6-y 66% PFS | 6-y 70% | 5 did not proceed to ASCT due to toxicity 1 MDS | 5 |
| ASCT | 60 | 95 (57) | 7 y EFS | 5-y 75% | 3 did not proceed to ASCT due to toxicity | 1.7 | |
| BR/RHiDAC29 | ASCT | 23 | 96 (96) | 1-y 96 | 1-y 100% | 0 | |
| Phase 3 | |||||||
| RCHOP vs RCHOP/RDHAP25 | ASCT | 455 | 98 vs 97 (63 vs 61) | 3.8-y PFS vs 7.3-y PFS | 6.8 y vs not reached | Hematologic and infectious | 4 |
| RDHAP28 | ASCT vs ASCT + MR | 299 | 4-y PFS 64% vs 83% | 4-y OS 89% vs 80% | 2 failed stem cell collection 38% received other platinum drug with first cycle | ||
| Transplant ineligible | |||||||
| Phase 3 | |||||||
| RCHOP vs RFC38 | NA | 455 | 86 vs 78 | TTF 28 mo vs 26 mo | 4-y 62% vs 47% | Primary hematologic | |
| 34 vs 40 | |||||||
| RCHOP vs BR40,41 | NA | 94 | 91 vs 93 | 22.1 vs 35.4 mo (median) | Median not reached | ||
| 30 vs 40 | Median not reached | ||||||
| RCHOP vs VRCAP43 | NA | 487 | 89 vs 92 (42 vs 53) | 14.4 vs 24.7 mo | 4-y 54% vs 64% | Primary hematologic | |
| Phase 2 | |||||||
| RBAC 50044 | NA | 57 | 91 | 2-y PFS 81% | Median not reached | Primary hematologic | |
| Nonchemotherapeutic | |||||||
| Rituximab + lenalidomide46,47 | LR | 38 | 92 | 4-y PFS 70% | 4-y 83% | Primary hematologic | |
| 64 | Rash; tumor flare | ||||||
| 7 secondary malignancies |
AML, acute myeloid leukemia; BR, bendamustine and rituximab; CRR, complete response rate; EFS, event-free survival; FC, rituximab, fludarabine, and cyclophosphamide; FFS, failure-free survival; LR, lenalidomide and rituximab; MDS, myelodysplastic syndrome; NA, not applicable; ORR, overall response rate; R-HyperCVAD, rituximab, fractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone alternating with high-dose methotrexate and cytarabine; RBAC, rituximab, bendamustine, and cytarabine; RBAC500, rituximab, bendamustine, and cytarabine; RHiDAC, rituximab and high-dose cytarabine; TRM, treatment-related mortality; RDHAP, rituximab, dexamethasone, cytarabine, and cisplatin; TTF, time to treatment failure, VR-CAP, cyclophosphamide, doxorubicin, bortezomib, and prednisone.
Younger/fit patients
Three separate phase 2 studies of rituximab, fractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone alternating with high-dose methotrexate and cytarabine (R-HyperCVAD) alternating with rituximab, high-dose methotrexate, and cytarabine (R-HCVAD/MA) established efficacy in younger patients.13-16 Investigators at M.D. Anderson reported high initial responses,13 recently reporting 15-year follow-up (median, 13.4 years) with median failure-free survival of 4.8 years and OS of 10.7 years, improving to 6.5 years and 13.4 years, respectively, in patients ≤65 years,17 although toxicity included myelodysplastic syndrome/acute myeloid leukemia (6.2%) in first remission. Two other multicenter studies were unsuccessful in replicating these results, reporting significant toxicity.14-16 A randomized phase 2 study conducted through the Southwest Oncology Group compared this regimen with bendamustine and rituximab (BR) prior to ASCT (S1106), but it was stopped early because of high failure of stem cell collection in the R-HyperCVAD/MA arm.18 Challenges with short- and long-term hematologic toxicity, plans to consolidate with ASCT, and patient age are limitations of this regimen. To minimize chemotherapy toxicity while incorporating the use of novel therapy, M.D. Anderson’s frontline trial of rituximab and ibrutinib (RI), followed by a shorter course of R-HyperCVAD/MA, enrolled 50 patients (NCT02427620).19 Patients received RI induction until best response, followed by a minimum of 4 chemotherapy cycles, reporting 100% overall response rate (ORR) and 80% complete response rate (CR) to RI induction. Although RI achieved impressive responses, all patients received aggressive chemotherapy consolidation; further follow-up is required for durability and cumulative toxicity.
The more commonly used approach in this population is consolidation with ASCT. Several induction regimens are effective prior to ASCT; the most used come from the Nordic Lymphoma Group (NLG) and the European MCL Network. The NLG established the importance of cytarabine in pretransplant induction with a phase 2 trial (MCL-2)20,21 of rituximab in combination with alternating cycles of maxi-CHOP (cyclophosphamide, doxorubicin, vincristine, prednisone) and high-dose cytarabine, followed by ASCT. This resulted in a 6-year PFS of 66% and a 6-year OS of 70%, a significant improvement in failure-free survival and OS over the NLG MCL-1 with intensified CHOP and ASCT.22 With 15-years of follow-up (median 11.4 years), median PFS was 8.5 years and OS was 12.7 years; however, late relapses were reported.23 Given similar follow-up, durable responses are comparable to that of R-HyperCVAD/MA,17 but with considerably less toxicity than combined data with R-HyperCVAD/MA and no plateau in OS. Based on a phase 2 French study evaluating RCHOP/RDHAP (rituximab, cisplatin, cytarabine, dexamethasone),24 the European MCL Network confirmed cytarabine’s role prior to ASCT25 by randomizing patients to RCHOP or RCHOP/RDHAP, followed by ASCT. After a median follow-up of 6.1 years, RCHOP/RDHAP resulted in significant improvement in TTF (9.1 vs 3.9 years, P = .038), although OS was not significantly different.26
Although the benefit of cytarabine is clear, the ideal regimen to maximize outcomes, while minimizing toxicity, is not. The NLG MCL5 trial evaluating rituximab in combination with high-dose cytarabine (RHDAC) was terminated early due to unacceptable rates of treatment failure.27 The Lymphoma Study Association evaluated RDHAP induction prior to transplant, eliminating the use of alkylating and anthracycline agents and limiting treatment to 4 cycles.28 RDHAP maintained high response rates and resulted in a promising 4-year PFS of 64% and 80% OS, but it has not been compared with RCHOP/RDHAP. A phase 2 study at Dana Farber Cancer Institute treated 23 patients with 3 cycles of BR, followed by 3 cycles of RHDAC, followed by ASCT, with 96% CR and 96% PFS at a median follow-up 13 months.29 Although promising, this study was small with a limited follow-up. A single-institution study at Washington University alternating BR with RHDAC is ongoing (NCT02728531). A larger multicenter study and longer follow-up will be needed to confirm efficacy and durability with this combination.
In the only randomized trial evaluating ASCT consolidation, the European MCL Network showed a PFS benefit with ASCT vs IFN-α maintenance after induction with a CHOP-like regimen,30 with an OS benefit seen at longer follow-up.31 Although this was prior to the incorporation of rituximab into induction therapy, a recently presented multicenter retrospective study of 1007 transplant-eligible MCL patients ≤65 years of age, in which 64% of patients received upfront ASCT32 and 94% received rituximab with induction, reported a PFS benefit (median, 44 vs 75 months, P < .01) and an OS benefit (median, 115 vs 147 months, P = .02) with ASCT at a median follow-up of 76.8 months (6.4 years).
With the benefits of MR established after chemo-immunotherapy in nontransplant regimens, the Lymphoma Study Association conducted a randomized phase 3 trial to evaluate the role of MR following ASCT in further improving response duration. Patients received RDHAP, followed by ASCT, and were randomized to observation vs MR every 8 weeks for 3 years.28 At a median follow-up of 50.2 months, 4-year event-free survival (79% vs 62%, P = .001), PFS (83% vs 64%, P < .001), and OS (89% vs 80%, P = .04) were significantly improved in patients receiving MR. While the benefits of MR have yet to be established with other induction regimens, MR should be considered after ASCT consolidation.
In an effort to explore the use of ibrutinib in frontline induction and maintenance and the role of ASCT with the inclusion of targeted therapy, The Triangle Study (EudraCT: 2014-001363-12) is an ongoing phase 3 trial being conducted by the European MCL Consortium. It randomized patients to 6 cycles of RCHOP/RDHAP, followed by ASCT; 6 cycles of RCHOP + ibrutinib/RDHAP, followed by ASCT and 2 years of ibrutinib maintenance (MI); or 6 cycles of RCHOP + ibrutinib/RDHAP and 2 years of MI (NCT02858258). It will be the first randomized study of ASCT in the era of rituximab and cytarabine induction.
Risk-stratified approach
The ability to achieve molecular remission is an independent predictor of outcome in MCL patients.33-36 Several trials have shown that achieving MRD negative remission in the peripheral blood and bone marrow, as measured by allele-specific oligonucleotide quantitative polymerase chain reaction, correlates with significant improvements in PFS, irrespective of regimen or treatment strategy.33-36 MRD positivity predicts clinical relapse, and the NLG has shown that preemptive treatment with rituximab, following the detection of MRD in patients previously MRD negative, could restore that MRD negativity, possibly delaying clinical relapse.35 The use of MRD to guide treatment decisions, including intensification or deintensification of therapy, treatment duration, maintenance approach, incorporation of targeted therapies, and preemptive treatment upon MRD detection, as opposed to clinical relapse, all need to be evaluated in prospective clinical trials; this will likely become easier with the utilization of next-generation sequencing to detect MRD.37 The Eastern Cooperative Oncology Group’s (ECOG-ACRIN) EA4151 is a phase 3 randomized study enrolling patients achieving MRD-negative remission after induction to ASCT + 3 years of MR vs 3 years of MR, irrespective of induction regimen (NCT03267433). This will assess the role of ASCT in the setting of achieving MRD negativity and is the first of likely many trials that will tailor treatment based on this approach.
Older patients
The majority of newly diagnosed patients are older, yet they are not candidates for intensive chemo-immunotherapy regimens or ASCT because of toxicities. The European MCL Network established RCHOP + MR as standard treatment for these patients in a 2-part randomized trial38 showing an OS benefit with RCHOP over R-FC (rituximab, fludarabine, cyclophosphamide) and a median remission duration significantly improved with MR until progression compared with IFN-α. Long-term follow-up (median, 6.7 years) confirmed prolonged PFS and OS with RCHOP + MR, with 5-year PFS of 51% and OS of 75%.39 Two randomized phase 3 trials established efficacy of BR, most convincingly the randomized phase 3 trial showing prolonged PFS with BR compared with RCHOP.40,41 Although there was no difference in OS, BR had an improved toxicity profile. Although MR is used after BR, its role is not established.42 Given bortezomib’s efficacy in relapsed disease, a randomized phase 3 trial evaluated R-CHOP vs cyclophosphamide, doxorubicin, bortezomib, and prednisone (VR-CAP), replacing vincristine with bortezomib.43 At a median follow-up of 40 months, VR-CAP had significantly improved PFS (24.7 vs 14.4 months, P < .001), although more hematologic toxicity was seen.
Given the efficacy and tolerability of BR, several studies aimed to improve upon this regimen. Recognizing the importance of cytarabine in younger patients, an Italian group evaluated BR with low-dose cytarabine.44 In a multicenter phase 2 trial, this combination resulted in 2-year PFS of 81%, although hematologic toxicity was considerable. Several studies incorporated nonchemotherapeutic agents with BR. A multicenter phase 1/2 trial evaluated BR + lenalidomide, followed by lenalidomide maintenance.45 At a median follow-up of 31 months, median PFS was 42 months, and 3-year OS was 73%. Infectious toxicity was high, and 8 (16%) patients developed a secondary malignancy, questioning the regimen’s tolerability. The results of 2 other frontline randomized studies are being anxiously awaited. ECOG’s E1411 phase 2 trial, designed to evaluate the addition of bortezomib to induction and the addition of lenalidomide to maintenance, randomized patients to 1 of 4 arms: (1) BR followed by MR, (2) BR followed by rituximab and lenalidomide maintenance (RL), (3) BR + bortezomib followed by MR, and (4) BR + bortezomib followed by RL (NCT01415752). The SHINE trial was a randomized phase 3 trial evaluating BR followed by MR vs BR + ibrutinib followed by rituximab + ibrutinib maintenance (NCT01776840). A randomized phase 3 trial evaluating BR vs BR + acalabrutinib is ongoing (NCT01972840). The benefit of BR combinations has been limited by increased toxicities, and these trial combinations will need to improve outcomes without producing unacceptable toxicity. The phase 3 MCL-R2 Elderly trial may address these concerns by evaluating the role of cytarabine-containing induction and lenalidomide-containing maintenance in older patients using RCHOP combinations. This trial randomizes to induction treatment with RCHOP vs RCHOP alternating with RHAD (rituximab, cytarabine, dexamethasone) with a second randomization to MR vs RL (2012-002542-20).
Nonchemotherapeutic approach
The goal of MCL therapy is to prolong disease remissions while minimizing toxicity, making nonchemotherapeutic approaches with active agents attractive. A multicenter phase 2 trial evaluated lenalidomide + rituximab induction, followed by RL maintenance, in 38 patients.46 This resulted in an impressive 2-year PFS and OS of 85% and 97%, respectively. The data were recently updated to show durable responses, with 4-year PFS and OS of 69.7% and 82.6%, respectively, and with 36% of patients remaining in remission beyond 5 years.47 Toxicity was primarily hematologic, and 7 secondary malignancies occurred: 5 skin cancers and 2 solid tumors. Although the numbers are small, this regimen was active across all risk groups, with response rates and durations comparable to chemotherapy. The efficacy of this 2-drug regimen should be compared in a randomized fashion with standard chemo-immunotherapy with MR. As discussed above, the combination of RI in treatment-naive patients induces very high response rates, although, in the setting of consolidation chemotherapy, the duration of response of RI alone is unknown, making its frontline role unclear.19 The triplet of rituximab + ibrutinib + lenalidomide (NCT03232307) is under evaluation in a phase 2 trial; if tolerable, it may represent an optimal approach.
Relapsed disease
Despite high response rates and improvement in PFS with current frontline approaches, patients will inevitably relapse. Management at relapse represents a unique challenge. Treatment choice is dependent on patient factors, prior therapy, remission duration, and candidacy for allogeneic stem cell transplant. Although an extensive review is not possible (Table 2), preferred approved therapy options at relapse include the Bruton’s tyrosine kinase inhibitors ibrutinib and acalabrutinib, with high interest in ibrutinib combinations. The approved agents bortezomib, lenalidomide, and temsirolimus have lower responses and response durations, providing limited utility as single agents and are best used in combination strategies.
Relapsed therapy agents and regimens
| Regimen | ORR (%) | CR (%) | PFS |
|---|---|---|---|
| Ibrutinib48-51 | 68-72 | 19-21 | 13-14.6 mo |
| Acalabrutinib57 | 81 | 40 | Median not reached |
| Lenalidomide68-71 | 28-40 | 5-12 | 4.0-8.8 mo |
| Bortezomib72,73 | 32-41 | 8-21 | 6.5 mo |
| Temsirolimus50,51,74 | 22-40 | 1-2 | 4.8-6.2 mo |
| RI58,59 | 88 | 44 | 43 mo |
| Lenalidomide + rituximab75 | 57 | 36 | 11.1 mo |
| Bortezomib + rituximab76 | 88 | 44 | 12.1 mo |
| Ibrutinib + lenalidomide + rituximab60 | 76 | 56 | Median not reached |
| Venetoclax62 | 75 | 21 | 14 mo |
| Venetoclax + ibrutinib64,65 | 71 | 71 | Median not reached, 12-mo PFS 75%, 18-mo PFS 57% |
| BR77-79 | 71-92 | 38-50 | 17.6-18.1 mo |
| RBAC80 | 80 | 70 | Median not reached, 2 y-PFS 70% |
| Bendamustine + bortezomib + rituximab81 | 71 | 52 | 2-y PFS 47% |
| Cyclophosphamide + doxorubicin + vincristine + prednisone82 | 47 | 21.7 | 8.1 mo |
| Cyclophosphamide + doxorubicin + vincristine + prednisone + bortezomib82 | 82.6 | 34.8 | 16.5 mo |
| Rituximab + gemcitabine + oxaliplatin83 | 83 | 60 | 22 mo |
| Gemcitabine + cisplatin + dexamethasone84 | 44 | 22 | 8.5 mo |
| Regimen | ORR (%) | CR (%) | PFS |
|---|---|---|---|
| Ibrutinib48-51 | 68-72 | 19-21 | 13-14.6 mo |
| Acalabrutinib57 | 81 | 40 | Median not reached |
| Lenalidomide68-71 | 28-40 | 5-12 | 4.0-8.8 mo |
| Bortezomib72,73 | 32-41 | 8-21 | 6.5 mo |
| Temsirolimus50,51,74 | 22-40 | 1-2 | 4.8-6.2 mo |
| RI58,59 | 88 | 44 | 43 mo |
| Lenalidomide + rituximab75 | 57 | 36 | 11.1 mo |
| Bortezomib + rituximab76 | 88 | 44 | 12.1 mo |
| Ibrutinib + lenalidomide + rituximab60 | 76 | 56 | Median not reached |
| Venetoclax62 | 75 | 21 | 14 mo |
| Venetoclax + ibrutinib64,65 | 71 | 71 | Median not reached, 12-mo PFS 75%, 18-mo PFS 57% |
| BR77-79 | 71-92 | 38-50 | 17.6-18.1 mo |
| RBAC80 | 80 | 70 | Median not reached, 2 y-PFS 70% |
| Bendamustine + bortezomib + rituximab81 | 71 | 52 | 2-y PFS 47% |
| Cyclophosphamide + doxorubicin + vincristine + prednisone82 | 47 | 21.7 | 8.1 mo |
| Cyclophosphamide + doxorubicin + vincristine + prednisone + bortezomib82 | 82.6 | 34.8 | 16.5 mo |
| Rituximab + gemcitabine + oxaliplatin83 | 83 | 60 | 22 mo |
| Gemcitabine + cisplatin + dexamethasone84 | 44 | 22 | 8.5 mo |
Ibrutinib became the first oral targeted agent to be approved by the US Food and Drug Administration (FDA) for MCL based on its unprecedented single-agent activity: 68% ORR, 21% CR, 13-month median PFS, and 22.5-month OS.48,49 Ibrutinib was well tolerated, but bleeding and atrial fibrillation emerged as potential serious adverse events.49 In a randomized phase 3 trial, ibrutinib had significantly better ORR, longer PFS, and better tolerability than temsirolimus,50 which were confirmed with longer follow-up.51 Evaluation of 370 patients treated in ibrutinib clinical trials with a median follow-up of 3.5 years showed that one third of patients received therapy for ≥2 years, with ∼10% on therapy for >4 years.52 Patients receiving ibrutinib at first relapse had a median PFS of 33.6 months, with median OS not reached, and a median DOR of 34.4 months, twice that of patients receiving >1 prior therapy and indicating the benefit of ibrutinib earlier in the treatment course. For patients achieving CR, median DOR was 4.5 years. In this cohort of patients, ECOG performance status, MIPI, bulky disease, and blastoid histology were associated with inferior PFS and OS.53 Patients with blastoid histology had similar ORR to those with nonblastoid histology but inferior DOR, PFS, and OS. Several retrospective studies have shown that patients progressing on ibrutinib have poor outcomes, regardless of subsequent therapy, with median survival < 1 year,54-56 although this likely is related, in part, to disease biology in heavily pretreated patients, with ibrutinib most beneficial in patients receiving 1 prior line of therapy.50-53 As ibrutinib routinely becomes used as first-line therapy at relapse, more effective therapies will be needed in those patients who subsequently relapse.
Acalabrutinib, a more selective Bruton’s tyrosine kinase inhibitor, was designed to minimize the off-target effects of ibrutinib in the hopes of reducing toxicities.57 Acalabrutinib received FDA approval for relapsed MCL in late October of 2017 based on a recently published phase 2 trial showing 81% ORR with 40% CR.57 With a median follow-up 15.2 months, median duration of response (DOR), PFS, and OS were not reached, with 12-month rates of 72%, 67%, and 87%, respectively. Compared with ibrutinib, patients were less heavily pretreated (median prior therapies, 2 vs 3), possibly contributing to the difference in ORR. Like ibrutinib, the majority of adverse events were grade 1-2; however, there were no reported cases of atrial fibrillation and 1 case (0.8%) of grade ≥3 hemorrhage. It remains to be seen whether improvement in these toxicities holds with more patients treated and longer follow-up. At this time, acalabrutinib may be preferred for patients at risk for cardiac toxicity or in need of anticoagulation or antiplatelet therapy.
Although ibrutinib has impressive responses, DOR is best in the limited patients achieving CR, with almost uniform development of resistance, at which time prognosis is poor. Trials evaluating ibrutinib combinations to improve depth of response and DOR are of high interest. The combination of IR in a single-center phase 2 trial achieved 88% ORR and 44% CR, comparing favorably to single-agent ibrutinib.58 The response rates were notably different in patients with Ki-67 levels < 50% vs ≥50%, with ORR of 100% and 50%, respectively. With further follow-up (median 47 months), 58% of patients achieved CR, and median PFS was 43 months.59 Patients with blastoid morphology, high-risk MIPI, and high Ki-67 had inferior survival, confirming the need for improved therapies in these high-risk patients. A multicenter phase 2 trial of the triplet ibrutinib, lenalidomide, and rituximab (PHILEMON) reported 76% ORR and 56% CR, with a median follow-up 17.8 months.60 Although the responses are promising, follow-up is limited, and 3 treatment-related deaths occurred, with more frequent toxicity, including neutropenia, infections, and cutaneous reactions. CDK4/6 inhibitors are active in MCL with preclinical data suggesting synergy with ibrutinib. A phase 1 study of ibrutinib + palbociclib had an acceptable safety profile, with 67% ORR and 44% CR, comparing favorably with studies of single-agent ibrutinib.61 A multicenter phase 2 study is planned (NCT078514). It will be critical moving forward to define therapy combinations with synergism and not just additive effects.
Newer agents
With multiple agents under investigation, the most promising therapies with potential for significant impact include the oral bcl-2 inhibitor venetoclax and chimeric antigen receptor (CAR) T cells.
Venetoclax has shown significant single-agent activity in 28 MCL patients, with 75% ORR, 21% CR, and median PFS of 14 months, comparing favorably with single-agent ibrutinib activity.62 Tumor lysis did occur with venetoclax treatment, and a ramp-up dosing schedule with close monitoring is required. Venetoclax and ibrutinib synergistically induce apoptosis in MCL cell lines63 providing rationale for the combination. Results from the phase 2 AIM study of the combination reported 71% ORR with 71% positron emission tomography CR.64,65 The MRD-negative rate was 67% by flow cytometry and 38% by allele-specific oligonucleotide quantitative polymerase chain reaction in all patients, increasing to 85% and 56%, respectively, in those with a detectable marker. At a median follow-up of 15.9 months, median PFS was not reached, with estimated 12-month and 18-month PFS of 75% and 57%, respectively. Responses were independent of MIPI and seen in 50% of patients with TP53 mutations or deletions; however, those with Ki-67 ≥ 30% were less likely to respond (P = .02). A phase 3 study of ibrutinib + placebo vs ibrutinib + venetoclax (NCT03112174) is ongoing. It remains to be seen whether the high CR and MRD rates with ibrutinib combinations translate into prolonged response durations. Other venetoclax combinations, including nonchemotherapeutic frontline options, are likely to emerge.
CAR-T cells are an adoptive immunotherapy approach demonstrating encouraging results in patients with relapsed/refractory B cell non-Hodgkin lymphoma, including few MCL patients, with recent FDA approval for relapsed large cell lymphoma.66,67 The ZUMA-2 trial (NCT02601313) is currently underway and is assessing the safety and efficacy of the autologous CD-19 CAR T-cell construct in patients with relapsed/refractory MCL failing ibrutinib. The role that CAR T cells will play in MCL is unclear, but they offer the potential of a curative approach in select patients outside of the limited role of allogeneic stem cell transplant.
Conclusions
The treatment of MCL has evolved with the use of rituximab- and cytarabine-containing regimens, ASCT consolidation in fit patients, and the use of maintenance rituximab in young and elderly patients. Despite improvements in remission durations, our current therapies are not providing a curative approach and are associated with acute and long-term toxicity. Several clinical and biologic markers are available to predict prognosis; however, despite improvements in outcomes, standard therapeutic approaches have not been able to overcome high-risk disease features. Incorporating targeted therapies, such as ibrutinib, into frontline therapy is desirable in an attempt to increase response durations and provide less-intensified combination therapies in hopes of sparing some toxicities; however, the success of new agents in achieving MRD negativity remains unknown. Nonchemotherapeutic approaches in the frontline and relapsed settings have become desirable based on the activity and limited toxicity of targeted agents, but they come with their own challenges: prolonged durations of treatment, chronic or cumulative toxicities, and financial burdens. It remains to be seen whether these therapies are able to overcome the adverse biology that eludes standard therapy. Relapse after ibrutinib is associated with a poor prognosis, and combination therapies to improve on the depth of response and DOR are needed, as are effective therapies for patients progressing after ibrutinib. CAR T cells provide a potential curative option; however, limited data exist in MCL and, with current toxicities, a majority of relapsed patients may not be candidates for this approach. Future trials should aim to deliver treatment based on disease behavior and biology, incorporate novel therapy combinations that induce deeper remissions, and use risk-stratification tools to tailor treatment approaches.
Authorship
Contribution: K.M. wrote the manuscript and generated the figure and tables.
Conflict-of-interest disclosure: K.M. has received research funding from Pharmacyclics, BMS, Merck, and Novartis and has consulted for Pharmacyclics, Janssen, Astra Zeneca, Genentech, BMS, and Seattle Genetics.
Correspondence: Kami Maddocks, Division of Hematology, Department of Internal Medicine, The Ohio State University Wexner Medical Center, Arthur G. James Comprehensive Cancer Center, 320 W 10th St, A347C Starling Loving Hall, Columbus, OH 43210; e-mail: kami.maddocks@osumc.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal