

TO THE EDITOR:
Mutations in the EGLN1 (also known as PHD2) gene are associated with erythrocytosis.1 The encoded protein, PHD2, is a central cellular oxygen sensor that hydroxylates the α subunit of the hypoxia inducible factor (HIF) transcription factor complex, marking it for degradation.2,-4 HIF controls red cell mass principally through upregulation of the ERYTHROPOIETIN (EPO) gene.5 All functionally characterized erythrocytosis-associated mutations in the EGLN1 gene described to date are heterozygous loss-of-function mutations that reside near or within the prolyl hydroxylase domain.6,,,,-11 However, PHD2 also contains a MYND-type zinc finger.12,13 Differing functions of this domain have been characterized, and it has been proposed to have either a positive or a negative regulatory function.14,-16 The importance of the zinc finger in humans is not known. Here, we report a human PHD2 zinc finger mutation associated with congenital erythrocytosis and its functional characterization. We find that this mutation produces a loss of function. This therefore supports the notion that the zinc finger ordinarily has a positive regulatory function and plays a role in the oxygen-sensing pathway that regulates erythropoiesis in humans.
The index case is a 14-year-old daughter of healthy consanguineous parents (first cousins) referred to a pediatrician because of persistent abdominal pain and fatigue. A general screening blood test revealed a hemoglobin (Hb) level of 18 g/dL (reference value 11.4-14.5 g/dL) (Table 1). Additional blood tests by a pediatric hematologist showed an elevated hematocrit Hct of 0.54 (reference value 0.37-0.45), RBC of 6.46 million/μL, WBC of 7.1 × 109/L, platelets of 217 × 109/L, and a low EPO of 6.0 mU/mL (reference value 9-28 mU/mL). Subsequent evaluation by a clinical geneticist led to whole exome sequencing as a first-tier routine diagnostic test (parent‐offspring trio approach). Whole exome sequencing was performed as described before17 and revealed a homozygous missense variant in the EGLN1 gene: EGLN1;Chr1(GRCh37):g.231557511A>G; NM_022051.2: c.124T>C (p.Cys42Arg). The mutated site shows strong amino-acid conservation (Grantham score: 180).
RBC measurements in a family with EGLN1-associated erythrocytosis
| Index | Brother | Father | Mother | |
|---|---|---|---|---|
| Hb, g/dL | 18 | 17.9 | 15.9 | 13.9 |
| (ref 11.4-14.5) | (ref 10.5-16.1) | (ref 13.7-17.7) | (ref 12.1-16.1) | |
| Hct | 0.54 | 0.52 | 0.48 | 0.44 |
| (ref 0.37-0.45) | (ref 0.35-0.50) | (ref 0.40-0.50) | (ref 0.40-0.50) | |
| RBC, million/μL | 6.46 | 6.4 | 5.8 | 4.4 |
| (ref 4.0-5.4) | (ref 3.8-5.6) | (ref 4.5-5.5) | (ref 4.0-5.0) | |
| EPO, mU/mL | 6 | 6.3 | 10 | 6.7 |
| (ref 9-28) | (ref 4.3-29) | (ref 4.3-29) | (ref 4.3-29) |
| Index | Brother | Father | Mother | |
|---|---|---|---|---|
| Hb, g/dL | 18 | 17.9 | 15.9 | 13.9 |
| (ref 11.4-14.5) | (ref 10.5-16.1) | (ref 13.7-17.7) | (ref 12.1-16.1) | |
| Hct | 0.54 | 0.52 | 0.48 | 0.44 |
| (ref 0.37-0.45) | (ref 0.35-0.50) | (ref 0.40-0.50) | (ref 0.40-0.50) | |
| RBC, million/μL | 6.46 | 6.4 | 5.8 | 4.4 |
| (ref 4.0-5.4) | (ref 3.8-5.6) | (ref 4.5-5.5) | (ref 4.0-5.0) | |
| EPO, mU/mL | 6 | 6.3 | 10 | 6.7 |
| (ref 9-28) | (ref 4.3-29) | (ref 4.3-29) | (ref 4.3-29) |
Hct, hematocrit; RBC, red blood cell; WBC, white blood cell.
Both parents were heterozygous for the variant. The brother of the index case was subsequently tested and found to be homozygous for the variant. His blood tests showed evidence of erythrocytosis with an Hb of 17.9 g/dL (reference value 10.5-16.1 g/dL), Hct of 0.52 (reference: 0.35-0.50), RBC of 6.4 million/μL (reference 3.8-5.6 million/μL), and an inappropriately normal EPO value. It may be noted that other EGLN1-linked erythrocytoses can be associated with normal, high, or even low EPO levels.18 The mother had normal Hb, Hct, RBC, and EPO levels, while the father had normal Hb, Hct, and EPO values, and a mildly elevated RBC at 5.8 million/μL (reference: 4.5-5.5 million/μL). All procedures followed were in accordance with the ethical standards of the institutional and national responsible committees on human experimentation and that, where relevant, informed consent was obtained.
The residue affected by the EGLN1 mutation, Cys-42, resides within the MYND-type zinc finger of PHD2 and is 1 of 8 evolutionarily conserved zinc-chelating residues (Figure 1A).12 We have previously reported that this zinc finger binds to a Pro-Xaa-Leu-Glu motif that is found within components of the HSP90 pathway, such as p23 and FKBP38.16 HIF-α is a client protein of the HSP90 pathway.19,20 Therefore, we have proposed that the zinc finger allows recruitment of PHD2 to the HSP90 pathway to facilitate prolyl hydroxylation of HIF-α.16
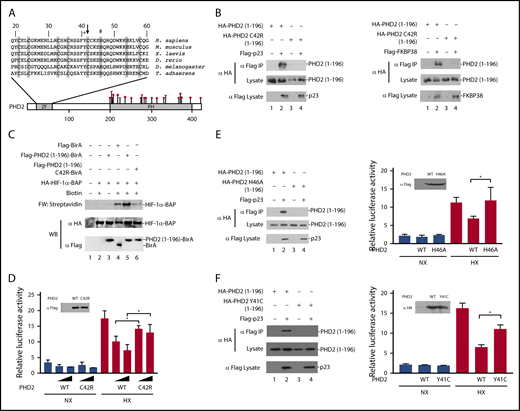
Functional studies on the effect of the C42R mutation on PHD2 activity. (A) Diagram of PHD2, showing locations of zinc finger (ZF) and prolyl hydroxylase (PH) domains. Sequence of the zinc finger across various metazoan species is shown at top. Shaded residues denote zinc-chelating residues. Arrow = Cys-42; + = Tyr-41; # = His-46. Previously reported erythrocytosis-associated mutations in PHD2 reside within or near the prolyl hydroxylase domain and comprise missense (circles), nonsense (diamond), and frameshift (triangle) mutations as shown. Numbers at top and bottom indicate residue number. (B) HEK293FT cells were transfected with constructs for the indicated proteins. Constructs were prepared by standard molecular biology techniques. Cells were lysed; the Flag-tagged proteins were immunoprecipitated, and the immunoprecipitates were examined for the absence or presence of hemagglutinin (HA)-tagged PHD2 (1-196) by anti-HA western blotting. Anti-HA and anti-Flag western blots of lysates are also shown. (C) In vitro transcription and translation reactions were performed for HA-HIF-1α BAP in the absence or presence of in vitro translated Flag-tagged BirA or BirA fusion proteins, as indicated. Biotinylation of HA-HIF-1α BAP was assessed by far-western (FW) blotting using streptavidin-alkaline phosphatase conjugates. Anti-HA and anti-Flag western blots (WB) are also shown. (D) HEK293FT cells in 96-well plates were transfected with 8 ng of (eHRE)3-Luc, 8 ng of RL-TK, and either 0.8 or 2.5 ng of either pcDNA3-Flag-PHD2 or pcDNA3-Flag-PHD2 C42R. DNA doses were held constant by the addition of pcDNA3. Eight hours after transfection, cells were exposed to 1% O2 (HX) or maintained under normoxia (NX) for an additional 16 hours. All cells were lysed, and luciferase activities were measured and normalized to that of the Renilla luciferase internal transfection control. Shown are means ± standard deviation, n = 3. *P < .05 by Student t test. Anti-Flag western blot of lysates of HEK293FT cells transfected with expression constructs for WT or C42R PHD2 is also shown. (E-F left panels) HEK293FT cells were transfected with constructs for the indicated proteins. Cells were lysed; the Flag-p23 was immunoprecipitated, and the immunoprecipitates were examined for the absence or presence of HA-tagged PHD2 (1-196) by anti-HA western blotting. Anti-HA and anti-Flag western blots of lysates are also shown. (E-F right panels) HEK293FT cells in 96-well plates were transfected with 8 ng of (eHRE)3-Luc, 8 ng of RL-TK, and 2.5 ng of either pcDNA3 or the indicated PHD2 expression constructs. Eight hours after transfection, cells were exposed to 1% O2 (HX) or maintained under normoxia (NX) for an additional 16 hours. All cells were lysed, and luciferase activities were measured and normalized to that of the Renilla luciferase internal transfection control. Shown are means ± standard deviation, n = 3-4. *P < .05 by Student t test. Western blots of lysates of HEK293FT cells transfected with PHD2 expression constructs are also shown.
Functional studies on the effect of the C42R mutation on PHD2 activity. (A) Diagram of PHD2, showing locations of zinc finger (ZF) and prolyl hydroxylase (PH) domains. Sequence of the zinc finger across various metazoan species is shown at top. Shaded residues denote zinc-chelating residues. Arrow = Cys-42; + = Tyr-41; # = His-46. Previously reported erythrocytosis-associated mutations in PHD2 reside within or near the prolyl hydroxylase domain and comprise missense (circles), nonsense (diamond), and frameshift (triangle) mutations as shown. Numbers at top and bottom indicate residue number. (B) HEK293FT cells were transfected with constructs for the indicated proteins. Constructs were prepared by standard molecular biology techniques. Cells were lysed; the Flag-tagged proteins were immunoprecipitated, and the immunoprecipitates were examined for the absence or presence of hemagglutinin (HA)-tagged PHD2 (1-196) by anti-HA western blotting. Anti-HA and anti-Flag western blots of lysates are also shown. (C) In vitro transcription and translation reactions were performed for HA-HIF-1α BAP in the absence or presence of in vitro translated Flag-tagged BirA or BirA fusion proteins, as indicated. Biotinylation of HA-HIF-1α BAP was assessed by far-western (FW) blotting using streptavidin-alkaline phosphatase conjugates. Anti-HA and anti-Flag western blots (WB) are also shown. (D) HEK293FT cells in 96-well plates were transfected with 8 ng of (eHRE)3-Luc, 8 ng of RL-TK, and either 0.8 or 2.5 ng of either pcDNA3-Flag-PHD2 or pcDNA3-Flag-PHD2 C42R. DNA doses were held constant by the addition of pcDNA3. Eight hours after transfection, cells were exposed to 1% O2 (HX) or maintained under normoxia (NX) for an additional 16 hours. All cells were lysed, and luciferase activities were measured and normalized to that of the Renilla luciferase internal transfection control. Shown are means ± standard deviation, n = 3. *P < .05 by Student t test. Anti-Flag western blot of lysates of HEK293FT cells transfected with expression constructs for WT or C42R PHD2 is also shown. (E-F left panels) HEK293FT cells were transfected with constructs for the indicated proteins. Cells were lysed; the Flag-p23 was immunoprecipitated, and the immunoprecipitates were examined for the absence or presence of HA-tagged PHD2 (1-196) by anti-HA western blotting. Anti-HA and anti-Flag western blots of lysates are also shown. (E-F right panels) HEK293FT cells in 96-well plates were transfected with 8 ng of (eHRE)3-Luc, 8 ng of RL-TK, and 2.5 ng of either pcDNA3 or the indicated PHD2 expression constructs. Eight hours after transfection, cells were exposed to 1% O2 (HX) or maintained under normoxia (NX) for an additional 16 hours. All cells were lysed, and luciferase activities were measured and normalized to that of the Renilla luciferase internal transfection control. Shown are means ± standard deviation, n = 3-4. *P < .05 by Student t test. Western blots of lysates of HEK293FT cells transfected with PHD2 expression constructs are also shown.
To examine whether the C42R PHD2 mutation affects PHD2 interaction with p23, we coexpressed wild-type or C42R PHD2 (residues 1-196) in HEK293FT cells, immunoprecipitated the p23, and examined the immunoprecipitates for the absence or presence of PHD2 using previously reported methods.16 As expected, wild-type PHD2 coimmunoprecipitates with p23 (Figure 1B left, top panel, lane 2). Importantly, the C42R mutation abolishes this interaction (Figure 1B left, top panel, lane 4). Similar results were obtained for the interaction between PHD2 and FKBP38 (Figure 1B right, top panel, lanes 2 and 4).
To examine whether these defective protein:protein interactions produce a functional defect, we employed an in vitro assay for PHD2 zinc finger function.21 In brief, we synthesized a fusion protein consisting of the PHD2 zinc finger fused to the birA biotin ligase from Escherichia coli. We added this protein to reactions in which we translated a HIF-α construct in which the primary site of prolyl hydroxylation is replaced by a biotin acceptor peptide (BAP) motif that can serve as a substrate for birA. BirA-catalyzed biotinylation of the BAP motif can be detected by far-western blotting using streptavidin (Figure 1C, top panel). The PHD2 zinc finger promotes recruitment of birA to the HSP90 pathway, which is present in the reticulocyte lysates employed in these assays. As reported previously,21 the level of biotinylation induced by the PHD2 zinc finger–birA fusion protein is significantly higher than that observed with birA alone (Figure 1C, top panel, lanes 4 and 5). Importantly, the C42R mutation abolishes this zinc finger–dependent enhancement of HIF-α biotinylation (Figure 1C, top panel, lane 6).
We next assessed the functional effect of the C42R PHD2 mutation in Hypoxia Response Element (HRE) reporter gene activity assays. We cotransfected HEK293FT cells with an HRE luciferase reporter gene and constructs for either wild-type or C42R PHD2. We exposed some cells to hypoxia (1% O2) and then measured reporter gene activity. As shown in Figure 1D, hypoxia activates the reporter gene, and this activity is decreased in a dose-dependent manner by wild-type PHD2 (columns 7 and 8). The mutant C42R PHD2 is significantly weaker than wild-type PHD2 in suppressing this hypoxia-induced reporter gene activity (columns 9 and 10).
To examine whether the functional defects observed are unique to Cys-42 in the zinc finger domain, we mutated a different zinc chelating residue, His-46 (Figure 1A). As with the C42R mutation, we find that the H46A mutation abolishes PHD2 binding to p23 and impairs its capacity to downregulate hypoxia-induced HRE activity (Figure 1E). Very recently, additional erythrocytosis-associated mutations in the zinc finger domain of PHD2 have been reported.22 Because these mutations were not functionally characterized, they were considered variants of unknown significance. We examined 1 of these, a Y41C mutation that changes a highly conserved residue (Figure 1A). As with the C42R and H46A mutations, the Y41C mutation abrogates PHD2 binding to p23 and produces a defect in its capacity to downregulate hypoxia-induced HRE activity (Figure 1F).
Collectively, the genetic and functional studies support the contention that the homozygous C42R mutation in the EGLN1 gene is the cause of erythrocytosis in the 2 affected siblings. We recently reported on the characterization of knock-in mice bearing a C36S/C42S mutation in the Egln1 allele that targets 2 of the predicted zinc chelating residues.21 One of these residues is, in fact, identical to that affected by the mutation reported here (Cys-42). As with the mice, the patients who are homozygous, but not heterozygous, for the C42R mutation display elevated Hb concentrations and Hcts. The requirement for 2 mutant alleles here could account for the rarity of this particular zinc finger mutation as opposed to catalytic domain mutations, the latter of which are sufficient to produce a phenotype in the heterozygous state.1 Importantly, the phenotype of the affected individuals reported here provides genetic evidence that the zinc finger of PHD2 normally plays a positive regulatory role in humans.
Acknowledgments
The authors thank the family that participated in this study.
This work was supported in part by National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases grant R01-DK104796 (F.S.L.) and National Institutes of Health, National Heart, Lung, and Blood Institute grant R33-HL120751 (F.S.L.). W.G. was supported by the Scientific Research Training Program for Young Talents (Union Hospital, Tongji Medical College, Huazhong University of Science and Technology).
Authorship
Contribution: M.S., D.S., W.G., J.W.H.J., B.E.N., K.P., A.E.D., and A.P.A.S. performed research and collected data; M.S., D.S., W.G., J.W.H.J., R.v.W., B.E.N., K.P., A.E.D., A.P.A.S., and F.S.L. analyzed data; and M.S., R.v.W., A.E.D., A.P.A.S., and F.S.L. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Margje Sinnema, Department of Clinical Genetics, Maastricht University Medical Center, P.O. Box 5800, 6202 AZ Maastricht, The Netherlands; e-mail: margje.sinnema@mumc.nl; and Frank S. Lee, Department of Pathology and Laboratory Medicine, Perelman School of Medicine, 605 Stellar Chance Labs, 422 Curie Blvd, Philadelphia, PA 19104; e-mail: franklee@pennmedicine.upenn.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal