

Key Points
A prospective reduced-intensity HCT trial for HLH/primary immunodeficiency resulted in low early mortality and 1-year OS of 80%.
Conditioning with fludarabine, melphalan, and alemtuzumab was associated with high rates of mixed chimerism and graft failure.

Abstract
Allogeneic hematopoietic cell transplantation (HCT) with myeloablative conditioning for disorders associated with excessive inflammation such as hemophagocytic lymphohistiocytosis (HLH) is associated with early mortality. A multicenter prospective phase 2 trial of reduced-intensity conditioning with melphalan, fludarabine, and intermediate-timing alemtuzumab was conducted for HLA matched or single HLA locus mismatched related or unrelated donor HCT in a largely pediatric cohort. Graft-versus-host disease (GVHD) prophylaxis was cyclosporine with methylprednisolone. The primary end point was 1-year overall survival (OS). Thirty-four patients with HLH and 12 with other primary immune deficiencies were transplanted. With a median follow-up of 20 months, the 1-year OS for transplanted patients was 80.4% (90% confidence interval [CI], 68.6%-88.2%). Five additional deaths by 16 months yielded an 18-month OS probability of 66.7% (90% CI, 52.9%-77.3%). Two patients experienced primary graft failure, and 18 patients either experienced a secondary graft failure or required a second intervention (mostly donor lymphocyte infusion [DLI]). At 1 year, the proportion of patients alive with sustained engraftment without DLI or second HCT was 39.1% (95% CI, 25.2%-54.6%), and that of being alive and engrafted (with or without DLI) was 60.9% (95% CI, 45.4 %-74.9%). The day 100 incidence of grade II to IV acute GVHD was 17.4% (95% CI, 8.1%-29.7%), and 1-year incidence of chronic GVHD was 26.7% (95% CI, 14.6%-40.4%). Although the trial demonstrated low early mortality, the majority of surviving patients required DLI or second HCT. These results demonstrate a need for future approaches that maintain low early mortality with improved sustained engraftment. The trial was registered at Clinical Trials.gov (NCT 01998633).
Introduction
Allogeneic hematopoietic cell transplant (HCT) is required for long-term survival for patients with several primary defects of the immune system. Although outcomes after related and unrelated donor HCT have improved in general in the last decades, survival after myeloablative (MAC) HCT in patients with diseases characterized by increased inflammation and immune dysregulation such as hemophagocytic lymphohistiocytosis (HLH) continues to be poor. The pediatric HLH-94 trial reported an overall 5-year survival of 50% for familial HLH (many died before HCT), with 66% of those who underwent HCT alive at 5 years.1 Other more recent studies reported similar outcomes with 3- to 5-year survival ranging from 49% to 64% with MAC conditioning approaches, with the vast majority of mortality occurring in the first 6 months after HCT.2-5 Some studies also note feasibility of umbilical cord transplant for HLH, with retrospective series reporting 65% to 71% long-term overall survival (OS).6,7
Shenoy et al reported a series of patients with a variety of nonmalignant conditions transplanted with a reduced-intensity conditioning (RIC) regimen that included alemtuzumab, fludarabine, and melphalan, with 12 of 14 patients surviving, including 1 of 2 patients with HLH.8 Subsequently, Cooper et al reported a single-center experience of 25 patients with HLH transplanted with a similar RIC approach. The researchers noted 85% OS (median, 36 months after HCT), with several patients disease-free with long-standing mixed chimerism.9 A strong case for potential superiority of RIC for HLH was made by a third report from the Cincinnati Children’s group, in which a retrospective review identified 43% estimated 3-year survival for patients transplanted with MAC (n = 14) compared with 92% for patients transplanted with RIC (n = 26).10
Other diseases with mechanisms related to dysfunctional immune mechanisms amenable to RIC HCT approaches include chronic active Epstein-Barr virus (EBV) disease (CAEBV); X-linked lymphoproliferative disease; Chediak-Higashi; chronic granulomatous disease (CGD); hyperimmunoglobulin M (HIGM1) syndrome; immune dysregulation, polyendocrinopathy, enteropathy, and X-linked (IPEX) syndrome; and leukocyte adhesion deficiency (LAD). CAEBV is characterized by highly elevated EBV levels in blood and tissue and is often associated with HLH, and case series suggest that HCT is required for survival for these patients.11 The other immune deficiencies included in this study share features of frequent comorbidities and preexisting infections. Registries and series reported OS similar to HLH after HCT: CAEBV (72%; median follow-up, 4 years),11 CGD (70%-100%; median follow-up, 17-61 months),12-18 HIGM1 (68%-90%; median follow-up, 1-3 years),19,20 IPEX (case reports),21 and LAD-1 (75%; median follow-up, 62 months).22
The improved survival reported for RIC strategies for HCT for HLH may be a result of the decreased toxicity of fludarabine/melphalan compared with myeloablative busulfan/cyclophosphamide. In addition, it is possible that alemtuzumab-mediated depletion of T cells and antigen-presenting cells ameliorates inflammation before HCT. Investigators from Cincinnati Children’s Hospital reported that the timing of alemtuzumab administration appeared to influence sustained engraftment, with 65% of the cohort developing mixed chimerism: 29% of patients who received “distal” alemtuzumab (starting day −22) and 79% of patients who received “proximal” alemtuzumab (start day range, −12 to −8). Most of the patients with mixed chimerism in this study ultimately received donor lymphocyte infusion (DLI) or second stem cell infusion.10 Mixed donor chimerism after RIC with alemtuzumab may be related to persistence of the drug at cytotoxic levels in patients after RIC for up to 60 days,23 with an estimated half-life of 15 to 21 days.24 Although a RIC approach for HLH appears to be effective, the optimal dose and timing of alemtuzumab and other therapeutic agents used in conditioning to achieve maximum survival with improved engraftment remained to be defined.
This multicenter prospective phase 2 trial, open to children and younger adults (4 months-45 years), aimed to test the safety and efficacy of an intermediate timing (day −14) of alemtuzumab as part of a RIC strategy for HLH and other primary immunodeficiencies (PIDs) (CAEBV, X-linked lymphoproliferative disease, Chediak-Higashi, CGD, HIGM1, IPEX, and LAD), where allogeneic HCT has been shown to be feasible and stable chimerism may be curative. Based on comparatively favorable engraftment in institutional series, intermediate timing strategy was tested in this study.10,25
Methods
Study design and conduct
This was a single-group, multicenter, phase 2 trial with a primary objective of estimating 1-year OS after transplantation with a RIC regimen consisting of alemtuzumab, fludarabine, and melphalan. Patients were followed on protocol for 1-year post-HCT, with additional follow-up available through the Center for International Blood and Marrow Transplant Research. The Institutional Review Board at each participating institution approved the study protocol, and parents, legal guardians, or patients aged 18 years or older provided informed consent for participation. An independent Data Safety Monitoring Board oversaw safety and conduct of the trial. The initial target sample size was 35 patients, but accrual was expanded to target 35 patients with HLH during an interim Data Safety Monitoring Board safety review. The trial was registered at Clinical Trials.gov (NCT 01998633). This manuscript was submitted on behalf of the Blood and Marrow Transplant Clinical Trials Network.
Patients
Patients aged 4 months through 45 years with HLH, CAEBV, CGD, HIGM1, IPEX syndrome, or LAD; performance score ≥50; and adequate cardiac, pulmonary, renal, and hepatic function were enrolled on trial. HLH encompassed patients meeting HLH-2004 diagnostic criteria for HLH requiring HCT for their disease, with the exclusion of malignancy-associated HLH.26 A detailed description of the inclusion criteria is shown in supplemental Table 1, available on the Blood Web site.
Exclusion criteria included prior HCT within 6 months of enrollment; administration of alemtuzumab within 2 weeks of enrollment; uncontrolled bacterial, viral, or fungal infection, with an exception for patients with CAEBV; history of prior or current malignancy, with the exception of EBV-associated lymphoma related to immune deficiency; seropositivity for HIV; or pregnant/breastfeeding women.
Donors
Donors were unaffected siblings matched at HLA-A and HLA-B (intermediate or higher resolution) and HLA-DRB1 (high-resolution DNA-based typing), or an unaffected relative or adult unrelated donor matched or mismatched at 1 HLA locus at HLA-A, HLA-B, HLA-C (intermediate or higher resolution), and HLA-DRB1 (high-resolution DNA-based typing). All donors must have been medically fit to donate bone marrow and met institutional (related donors) or National Marrow Donor Program (unrelated donors) criteria for donation.
Treatment
The transplant-conditioning regimen included alemtuzumab on days −14 through −10 (1 mg/kg subcutaneous with a maximum cumulative dose of 90 mg), fludarabine on days −8 through −4 (150 mg/m2 or 5 mg/kg in patients weighing <10 kg), and melphalan on day −3 (140 mg/m2 or 4.7 mg/kg in patients weighing <10 kg). Bone marrow was the specified graft and was infused on day 0 per local institutional guidelines. GVHD prophylaxis consisted of cyclosporine (day −3 through day +100) tapered through day +180 and methylprednisolone 2 mg/kg/day (day −2 and −1), 1 mg/kg/day through day+28, and then tapered over a month. Oral prednisone (1.2 mg/kg/day) may have been substituted after HCT. Supportive care recommendations included prophylaxis and surveillance for viral and fungal infection, prophylaxis for Pneumocystsis jirovecii and fungal infections, strict blood pressure monitoring, and prompt treatment of suspected or overt infections. The complete protocol is included in supplemental Materials (supplemental Appendix A). Guidelines and recommendations for infection prophylaxis and surveillance are outlined in Protocol Section 2.7.6. Granulocyte colony-stimulating factor was not required, but could be given at provider discretion. Chimerism studies (overall and T cell) were required monthly after engraftment, day +100, day +180, day +365. Recommendations for managing chimerism and graft loss were included in Protocol Section 2.6.
Outcomes
The primary end point was 1-year OS. All other outcomes studied were considered secondary. Sustained engraftment was defined as whole-blood donor chimerism higher than 5% by day +42 and continued maintenance of donor chimerism higher than 5% through the first year after transplantation. Donor chimerism of 5% or less, a decline in donor chimerism to less than 5% after initial engraftment, DLI, or second transplant (with or without conditioning) were all considered failures of sustained engraftment.27 Neutrophil recovery was defined as the first of 3 days when the absolute neutrophil count was at least 0.5 × 109/L. Platelet recovery was defined as the first of 7 days without platelet transfusion that the platelet count was at least 20 × 109/L. Acute and chronic GVHD were graded using standard criteria.28,29 Lymphocyte subset analysis, immunoglobulin (Ig) levels, and NK cell function were performed with clinical assays. Performance status was determined with an age-appropriate scale (Lansky, <18; Karnofsky, ≥18).
Disease reactivation was studied for HLH and CAEBV only. HLH reactivation was defined by clinical and laboratory evidence of pathologic inflammation (persistent fever, progressive cytopenia or cytopenias, rising ferritin and soluble IL2Rα, decreasing fibrinogen, hepatosplenomegaly, end-organ damage) not attributable to other causes. Reactivation of central nervous system (CNS) HLH was defined by pleocytosis in cerebrospinal fluid or magnetic resonance imaging consistent with CNS inflammation not attributable to other causes. CAEBV reactivation was defined as detection of recipient EBV-positive lymphocytes in lymphocytes or tissue. Common Terminology Criteria for Adverse Events version 3.0 was used to report expected and unexpected grade 3 to 5 adverse events.
Statistical analysis
The primary objective was to estimate 1-year OS after HCT with a RIC regimen with melphalan, fludarabine, and intermediate timing of alemtuzumab (day −14 to day −10). The trial was designed such that an observed OS of 90% would rule out survival percentages below 70%, based on a 90% confidence interval (CI). During the course of the study, the Data Safety Monitoring Board allowed an increase in accrual to target a larger number of patients with HLH. OS was estimated using the Kaplan-Meier estimator, and the 90% CI was calculated using the Greenwood formula. Frequency and proportion of sustained engraftment was estimated, along with an associated exact 95% Clopper-Pearson confidence interval.30 The incidence of neutrophil and platelet recovery and acute and chronic GVHD were estimated using the cumulative incidence estimator to accommodate death as the only the competing risk for these events.31 Recovery of immune system assessed by immune assays posttransplant was reported using summary statistics, and changes from baseline were tested using a Wilcoxon Signed Rank and a Benjamini-Hochberg P-value adjustment.32 Similarly, a description of major regimen-related toxicities, infections, and causes of death was reported. Prespecified disease-specific subgroup analysis (HLH and other PID) was also conducted using similar methods. Analyses includes data collected as of April 2017. Analyses were performed using SAS v9.4 and R v3.2.
Results
Patients and transplant characteristics
The trial opened in November 2013 and closed to accrual in December 2015. Twenty-two sites enrolled 47 patients. The median follow-up time for surviving patients was 597 days (range, 351-811 days). One patient with HLH died after enrollment but before initiating the conditioning regimen and was excluded from analysis, as specified in the protocol. The characteristics of the 46 patients who underwent HCT are shown in Table 1: 34 patients had HLH and 12 patients had other primary immune deficiency diseases. In this study, 25 (69%) of 36 transplanted HLH participants reported an inherited gene mutation associated with HLH and 9 (25%) of 36 met clinical criteria for HLH without reported gene defects. The median age at HCT was 2.3 years (range, 5 months-28 years), and 34 patients (74% of all transplanted patients) were less than 10 years old. Thirty-five patients (76%) were Caucasian, and 15 (33%) were female. Thirty-four (74%) reported a performance score of 90 or 100 at HCT, 8 (17%) a performance score of 80, 2 (4%) a performance score of 70, and the remaining 2 (4%) a performance score of 60 or 50. The indication for HCT was HLH for 34 (74%), CAEBV for 3 (7%), CGD for 5 (11%), HIGM1 for 2 (4%), and IPEX for 2 (4%) of patients. Although eligible, there were no patients with LAD enrolled on study. The predominant donor was an HLA-matched unrelated donor (n = 25; 54%) or 1 HLA-locus mismatched unrelated donor (n = 13; 28%). Only 7 patients (15%) received their graft from an HLA-matched sibling, and 1 patient (2%) received a graft from a mismatched related donor. One patient received peripheral blood instead of bone marrow because the donor was declared unfit for marrow donation.
Patient, disease and transplant characteristics
| Primary disease | All diseases, n (%)* | ||
|---|---|---|---|
| HLH, n (%)* | Other PID, n (%)* | ||
| Age, y | |||
| <1 | 13 (38) | 1 (8) | 14 (30) |
| 1-9 | 13 (38) | 7 (58) | 20 (43) |
| 10-19 | 7 (21) | 2 (17) | 9 (20) |
| 20-29 | 1 (3) | 2 (17) | 3 (7) |
| Sex | |||
| Female | 11 (32) | 4 (33) | 15 (33) |
| Male | 23 (68) | 8 (67) | 31 (67) |
| Race | |||
| Caucasian | 26 (76) | 9 (75) | 35 (76) |
| African American | 4 (12) | 2 (17) | 6 (13) |
| Asian | 2 (6) | 0 (0) | 2 (4) |
| Not reported | 2 (6) | 1 (8) | 3 (7) |
| Performance score | |||
| 90-100 | 24 (70) | 10 (83) | 34 (74) |
| 80 | 6 (18) | 2 (17) | 8 (17) |
| 70 | 2 (6) | 0 (0) | 2 (4) |
| 60 | 1 (3) | 0 (0) | 1 (2) |
| 50 | 1 (3) | 0 (0) | 1 (2) |
| Disease type | |||
| Hemophagocytic lymphohistiocytosis | 34 (100) | 0 (0) | 34 (74) |
| Chronic active Epstein-Barr virus | 0 (0) | 3 (25) | 3 (7) |
| Chronic granulomatous disease | 0 (0) | 5 (42) | 5 (11) |
| HIGM1 syndrome | 0 (0) | 2 (17) | 2 (4) |
| IPEX | 0 (0) | 2 (17) | 2 (4) |
| Donor type | |||
| HLA-matched sibling | 7 (21) | 0 (0) | 7 (16) |
| 1 HLA locus mismatched relative | 0 (0) | 1 (8) | 1 (2) |
| HLA matched unrelated donor | 16 (47) | 9 (75) | 25 (54) |
| 1 HLA locus mismatched unrelated donor | 11 (32) | 2 (17) | 13 (28) |
| Primary disease | All diseases, n (%)* | ||
|---|---|---|---|
| HLH, n (%)* | Other PID, n (%)* | ||
| Age, y | |||
| <1 | 13 (38) | 1 (8) | 14 (30) |
| 1-9 | 13 (38) | 7 (58) | 20 (43) |
| 10-19 | 7 (21) | 2 (17) | 9 (20) |
| 20-29 | 1 (3) | 2 (17) | 3 (7) |
| Sex | |||
| Female | 11 (32) | 4 (33) | 15 (33) |
| Male | 23 (68) | 8 (67) | 31 (67) |
| Race | |||
| Caucasian | 26 (76) | 9 (75) | 35 (76) |
| African American | 4 (12) | 2 (17) | 6 (13) |
| Asian | 2 (6) | 0 (0) | 2 (4) |
| Not reported | 2 (6) | 1 (8) | 3 (7) |
| Performance score | |||
| 90-100 | 24 (70) | 10 (83) | 34 (74) |
| 80 | 6 (18) | 2 (17) | 8 (17) |
| 70 | 2 (6) | 0 (0) | 2 (4) |
| 60 | 1 (3) | 0 (0) | 1 (2) |
| 50 | 1 (3) | 0 (0) | 1 (2) |
| Disease type | |||
| Hemophagocytic lymphohistiocytosis | 34 (100) | 0 (0) | 34 (74) |
| Chronic active Epstein-Barr virus | 0 (0) | 3 (25) | 3 (7) |
| Chronic granulomatous disease | 0 (0) | 5 (42) | 5 (11) |
| HIGM1 syndrome | 0 (0) | 2 (17) | 2 (4) |
| IPEX | 0 (0) | 2 (17) | 2 (4) |
| Donor type | |||
| HLA-matched sibling | 7 (21) | 0 (0) | 7 (16) |
| 1 HLA locus mismatched relative | 0 (0) | 1 (8) | 1 (2) |
| HLA matched unrelated donor | 16 (47) | 9 (75) | 25 (54) |
| 1 HLA locus mismatched unrelated donor | 11 (32) | 2 (17) | 13 (28) |
Column percentage given.
Overall survival
There were 9 deaths within the first year after HCT, and 1-year OS for the whole cohort was 80.4% (90% CI, 68.6%-88.2%; Figure 1A). There were 5 additional deaths by 16 months after HCT. With a total of 14 deaths at last follow-up, the probability of 18-month OS for the whole cohort was 66.7% (90% CI, 52.9%-77.3%). Disease-specific OS for HLH and other primary immune deficiency diseases are shown in Figure 1B. The 1-year and 18-month rates of OS for HLH were 82.4% (90% CI, 68.4%-90.6%) and 68% (90% CI, 51.8%-79.7%), respectively. The corresponding rates of OS for other immune deficiency diseases were 75% (90% CI, 47.4%-89.5%) and 62.5% (90% CI, 32.7%-82.0%). Of the 14 deaths, 1 was attributed to disease recurrence (T-cell posttransplant lymphoproliferative disease in a patient with CAEBV), 3 to infections (adenovirus, fungal, and Enterobacter), 4 to acute GVHD, 3 to organ failure, 1 to secondary malignancy (T-cell posttransplant lymphoproliferative disease in a patient with HLH), 1 to autoimmune hemolytic anemia, and 1 to autoimmune thrombocytopenia after second transplantation (Table 2).
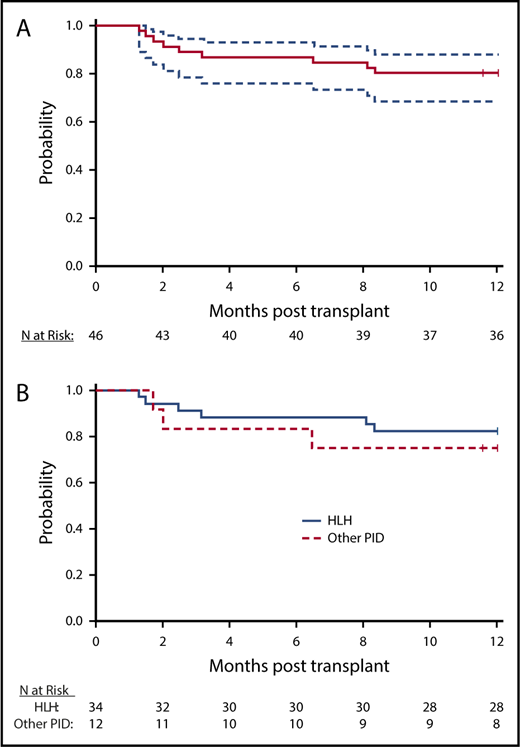
OS. (A) Survival curve demonstrates OS (solid line) with 95% CIs (dashed lines). (B) HLH and non-HLH survival. Survival curve illustrates probability of survival for HLH (blue) and subjects with other PID (red).
OS. (A) Survival curve demonstrates OS (solid line) with 95% CIs (dashed lines). (B) HLH and non-HLH survival. Survival curve illustrates probability of survival for HLH (blue) and subjects with other PID (red).
Causes of death
| Primary cause of death | HLH participants, n (%) | Other PID participants, n (%) | Transplanted participants, n (%) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| All | Before 1 y | After 1 y | All | Before 1 y | After 1 y | All | Before 1 y | After 1 y | |
| Acute GVHD* | 2 (20) | 1 (10) | 1 (10)† | 2 (50) | 1 (25) | 1 (25) | 4 (28.6) | 2 (14.3) | 2 (14.3) |
| Infection | 3 (30) | 2 (20) | 1 (10) | 0 (0) | 0 (0) | 0 (0) | 3 (21.4) | 2 (14.3) | 1 (7.1) |
| Multiorgan failure | 1 (10) | 1 (10) | 0 (0) | 1 (25) | 1 (25) | 0 (0) | 2 (14.3) | 2 (14.3) | 0 (0) |
| Organ failure | 1 (10) | 1 (10) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (7.1) | 1 (7.1) | 0 (0) |
| Primary disease recurrence | 0 (0) | 0 (0) | 0 (0) | 1 (25) | 1 (25) | 0 (0) | 1 (7.1) | 1 (7.1) | 0 (0) |
| Second malignancy | 1 (10) | 1 (10) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (7.1) | 1 (7.1) | 0 (0) |
| Other | 2 (20) | 0 (0) | 2 (20) | 0 (0) | 0 (0) | 0 (0) | 2 (14.3) | 0 (0) | 2 (14.3) |
| All causes | 10 (100) | 6 (60) | 4 (40) | 4 (100) | 3 (75) | 1 (25) | 14 (100) | 9 (64.3) | 5 (35.7) |
| Primary cause of death | HLH participants, n (%) | Other PID participants, n (%) | Transplanted participants, n (%) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| All | Before 1 y | After 1 y | All | Before 1 y | After 1 y | All | Before 1 y | After 1 y | |
| Acute GVHD* | 2 (20) | 1 (10) | 1 (10)† | 2 (50) | 1 (25) | 1 (25) | 4 (28.6) | 2 (14.3) | 2 (14.3) |
| Infection | 3 (30) | 2 (20) | 1 (10) | 0 (0) | 0 (0) | 0 (0) | 3 (21.4) | 2 (14.3) | 1 (7.1) |
| Multiorgan failure | 1 (10) | 1 (10) | 0 (0) | 1 (25) | 1 (25) | 0 (0) | 2 (14.3) | 2 (14.3) | 0 (0) |
| Organ failure | 1 (10) | 1 (10) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (7.1) | 1 (7.1) | 0 (0) |
| Primary disease recurrence | 0 (0) | 0 (0) | 0 (0) | 1 (25) | 1 (25) | 0 (0) | 1 (7.1) | 1 (7.1) | 0 (0) |
| Second malignancy | 1 (10) | 1 (10) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (7.1) | 1 (7.1) | 0 (0) |
| Other | 2 (20) | 0 (0) | 2 (20) | 0 (0) | 0 (0) | 0 (0) | 2 (14.3) | 0 (0) | 2 (14.3) |
| All causes | 10 (100) | 6 (60) | 4 (40) | 4 (100) | 3 (75) | 1 (25) | 14 (100) | 9 (64.3) | 5 (35.7) |
Deaths through last contact date, including those that occurred after 1 year. Percentage of the deaths within each disease category is given.
One HLH participant died on day 482 as a result of acute GVHD according to Center for International Blood and Marrow Transplant Research records; cause of death was not adjudicated by end point review committee
Disease recurrence was studied for HLH and CAEBV. Of the 34 patients with HLH, 5 patients experienced reactivation, including 1 with 2 separate reactivations, and the incidence of HLH reactivation at 1 year was 14.7% (95% CI, 5.2%-28.7%). Among the 3 patients with CAEBV, 1 died of disease reactivation at day 61, 1 maintained negative EBV qPCR after day 100, and 1 redeveloped clinically insignificant detectable EBV after day 100.
Hematopoietic recovery and engraftment
All patients achieved neutrophil recovery. The median time to neutrophil recovery was 13 days, and the day 42 incidence of neutrophil recovery was 100% (95% CI, 90.8%-100.0%). The day 100 incidence of platelet recovery was 88.9% (95% CI, 73.3%-95.6%). All patients who recovered platelets 20 × 109/L by day 100 also recovered platelets 50 × 109/L by day 100. Two patients with neutrophil recovery had less than 5% donor chimerism through day 42 post-HCT, and 1 patient with 53% donor chimerism at day 28 died on day 39. The proportion of patients who were alive with neutrophil recovery and donor chimerism higher than 5% at day 42 was 93.5% (95% CI, 82.1%-98.6%). Beyond day 42, 26 patients had sustained donor chimerism higher than 5% without an intervention at last follow-up, of which 18 were alive at 1 year and 8 died before 1 year after HCT. The remaining 17 patients experienced a decline in donor chimerism to less than 5% (n = 7) or required DLI or second HCT (n = 10) for declining donor chimerism, with the treatment intervention occurring before attaining less than 5% donor chimerism. At 1 year, the estimated probability of being alive with sustained engraftment (without an intervention) was 39.1% (n = 18; 95% CI, 25.1%-54.6%), and that of being alive and engrafted (with or without an intervention) was 60.9% (n = 28; 95% CI, 45.4%-74.9%; Figure 2A-B; Table 3). The median chimerism (unfractionated) for all subjects with sustained engraftment at 1 year was 99% (range, 23%-100%); 92.9% for HLH (range, 23%-100%), and 94.5% for other PID (range, 80%-100%). The proportion of patients with HLH who were alive with sustained engraftment (without an intervention) was 41.2% (n = 14; 95% CI, 24.7%-59.3%), and that for other primary immune diseases was 33.3% (n = 4; 95% CI, 9.9%-65.1%). The chimerism (unfractionated or T cell) for patients alive and without intervention at 1 year and for surviving patients who required DLI or second transplant for sustained engraftment are shown in Table 4 and supplemental Table 2. These data were not reported at 365 days for 4 patients; 1 reported 100% donor cells at day 100, 1 only had fractionated samples available, 1 was lost to follow-up before the 1-year visit, and 1 required a second infusion before the 1-year visit and was not assessed for chimerism after that. The cumulative incidence of administration of additional cells products (DLI or 2nd HCT) at 1 year was 41.1% (95% CI, 26.9%-55.3%; treating death as a competing risk). Interestingly, in 4 cases in which serial chimerism data were available, T-cell and/or whole-cell chimerism was worse after the DLI (supplemental Table 2).
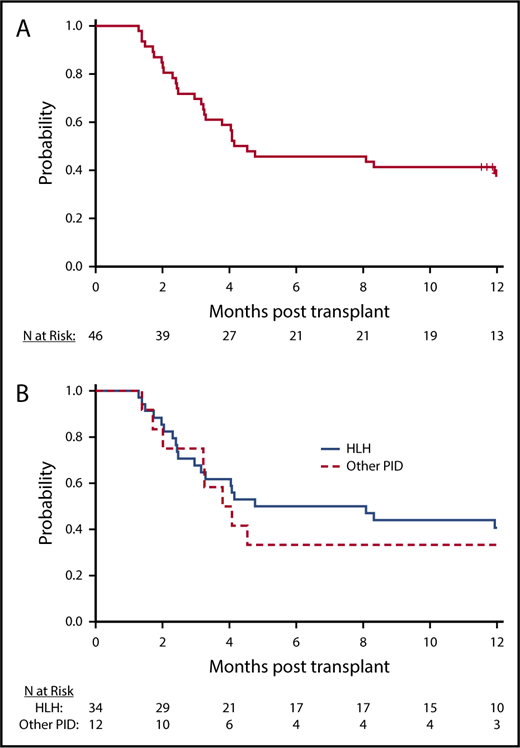
Engraftment. (A) Probability of intervention-free engraftment, all transplanted patients. Kaplan-Meier curve illustrates probability of sustained engraftment without intervention. (B) Probability of intervention-free engraftment, by primary disease. Kaplan-Meier curve illustrates probability of sustained engraftment without intervention by primary disease (solid line, HLH; dashed line, other PID).
Engraftment. (A) Probability of intervention-free engraftment, all transplanted patients. Kaplan-Meier curve illustrates probability of sustained engraftment without intervention. (B) Probability of intervention-free engraftment, by primary disease. Kaplan-Meier curve illustrates probability of sustained engraftment without intervention by primary disease (solid line, HLH; dashed line, other PID).
Engraftment status
| Primary disease | All diseases, n (%) | ||
|---|---|---|---|
| HLH, n (%) | Other PID, n (%) | ||
| Total (n) | 34 | 12 | 46 |
| Sustained engraftment | |||
| Sustained donor chimerism >5%, alive at 1 y, no intervention | 14 (41) | 4 (33) | 18 (39) |
| Received DLI and/or second HCT | |||
| Donor chimerism >5% after intervention | 9 (26) | 1 (8) | 10 (22) |
| Donor chimerism ≤5% after intervention | 3 (9) | 2 (17) | 5 (11) |
| Primary graft failure | |||
| Donor chimerism remained ≤5% after initial HCT | 1 (3) | 1 (8) | 2 (4) |
| Secondary graft failure | |||
| Donor chimerism ≤5% without intervention | 1 (3) | 1 (8) | 2 (4) |
| Not evaluable for sustained engraftment | |||
| Donor chimerism 53% at day 28, died before day 42 | 1 (3) | 0 (0) | 1 (2) |
| Sustained donor chimerism >5%, dead before 1 y | 5 (15) | 3 (25) | 8 (17) |
| Primary disease | All diseases, n (%) | ||
|---|---|---|---|
| HLH, n (%) | Other PID, n (%) | ||
| Total (n) | 34 | 12 | 46 |
| Sustained engraftment | |||
| Sustained donor chimerism >5%, alive at 1 y, no intervention | 14 (41) | 4 (33) | 18 (39) |
| Received DLI and/or second HCT | |||
| Donor chimerism >5% after intervention | 9 (26) | 1 (8) | 10 (22) |
| Donor chimerism ≤5% after intervention | 3 (9) | 2 (17) | 5 (11) |
| Primary graft failure | |||
| Donor chimerism remained ≤5% after initial HCT | 1 (3) | 1 (8) | 2 (4) |
| Secondary graft failure | |||
| Donor chimerism ≤5% without intervention | 1 (3) | 1 (8) | 2 (4) |
| Not evaluable for sustained engraftment | |||
| Donor chimerism 53% at day 28, died before day 42 | 1 (3) | 0 (0) | 1 (2) |
| Sustained donor chimerism >5%, dead before 1 y | 5 (15) | 3 (25) | 8 (17) |
Last available chimerism data before 1 year, death, graft failure, DLI, and/or second transplant
| ID | Primary diagnosis | Sustained engraftment or failure event | Day of 1-year follow-up or failure event | Last available chimerism | Minimum recorded chimersim | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Day of unfractionated sample | Unfractionated sample, % donor | Unfractionated sample type | Day of T-cell sample | T-cell sample, % donor | T-cell sample type | % Donor | Day of sample | Sample type | ||||
| 1 | CAEBV | Sustained engraftment | 365 | — | — | — | 102 | 100 | Blood | 69 | 12 | T-cell |
| 2 | HLH | Sustained engraftment | 365 | — | — | — | 124 | 100 | Blood | 85 | 26 | T-cell |
| 3 | HLH | Death | 39 | 19 | 99 | Blood | 37 | 53 | Blood | 53 | 37 | T-cell |
| 4 | HLH | Sustained engraftment | 364 | 144 | 100 | Blood | 144 | 100 | Blood | 95 | 25 | T-cell |
| 5 | HLH | DLI | 53 | 39 | 54 | Blood | — | — | — | 4 | 18 | T-cell |
| 6 | HIGM1 | DLI | 115 | — | — | — | 98 | 97 | Blood | 92 | 76 | Marrow |
| 7 | HLH | DLI | 60 | 52 | 55 | Blood | — | — | — | 55 | 52 | Marrow |
| 8 | HLH | DLI | 73 | 70 | 24 | Blood | 70 | 37 | Blood | 2 | 170 | Marrow |
| 9 | HLH | Sustained engraftment | 365 | — | — | — | 354 | 95 | Blood | 89 | 35 | T-cell |
| 10 | HLH | DLI | 90 | 83 | 81 | Blood | 76 | 63 | Blood | 21 | 356 | Marrow |
| 11 | HLH | DLI | 74 | 73 | 28 | Blood | 73 | 82 | Blood | 5 | 307 | Marrow |
| 12 | HLH | SGF | 70 | 54 | 38 | Blood | 54 | 14 | Blood | 4 | 70 | T-cell |
| 13 | HLH | DLI | 100 | 84 | 34 | Blood | 84 | 89 | Blood | 32 | 68 | Marrow |
| 14 | HLH | Sustained engraftment | 365 | — | — | — | 245 | 95 | Blood | 93 | 175 | T-cell |
| 15 | HLH | DLI | 126 | 101 | 64 | Blood | 101 | 57 | Blood | 7 | 318 | Marrow |
| 16 | HLH | DLI | 123 | 88 | 30 | Blood | 88 | 14 | Blood | 12 | 53 | T-cell |
| 17 | HLH | Death | 253 | 248 | 100 | Blood | — | — | 100 | 24 | Marrow | |
| 18 | CAEBV | Sustained engraftment | 365 | — | — | — | 361 | 80 | Blood | 31 | 46 | T-cell |
| 19 | HLH | Death | 45 | 39 | 96 | Blood | 39 | 96 | Blood | 96 | 27 | Marrow |
| 20 | HLH | Sustained engraftment | 361 | — | — | — | 188 | 99 | Blood | 56 | 39 | T-cell |
| 21 | HLH | PGF | 42 | 41 | 0 | Blood | 41 | 0 | Blood | 0 | 27 | Marrow |
| 22 | IPEX | DLI | 138 | 105 | 15 | Blood | — | — | — | 8 | 163 | Marrow |
| 23 | HLH | Sustained engraftment | 365 | 175 | 98 | Blood | — | — | — | 97 | 371 | Marrow |
| 24 | HLH | Sustained engraftment | 363 | 333 | 90 | Blood | 333 | 97 | Blood | 90 | 27 | T-cell |
| 25 | HLH | Sustained engraftment | 365 | 275 | 100 | Blood | — | — | — | 100 | 16 | Marrow |
| 26 | CGD | SGF | 98 | — | — | — | 85 | 18 | Blood | 0 | 106 | T-cell |
| 27 | CGD | DLI | 124 | 89 | 5 | Blood | 89 | 10 | Blood | 4 | 152 | Marrow |
| 28 | HLH | Death | 96 | 84 | 100 | Blood | — | — | — | 100 | 28 | Marrow |
| 29 | HLH | Sustained engraftment | 365 | — | — | — | 362 | 100 | Blood | 100 | 21 | T-cell |
| 30 | HLH | Sustained engraftment | 365 | 312 | 100 | Blood | — | — | — | 96 | 84 | Marrow |
| 31 | HLH | Death | 246 | 243 | 99 | Blood | — | — | — | 67 | 26 | Marrow |
| 32 | CGD | Sustained engraftment | 351 | 169 | 96 | Blood | 169 | 96 | Blood | 96 | 56 | Marrow |
| 33 | HLH | Sustained engraftment | 356 | 342 | 23 | Blood | 342 | 79 | Blood | 23 | 342 | Marrow |
| 34 | CGD | Sustained engraftment | 365 | 70 | 93 | Blood | — | — | — | 93 | 70 | Marrow |
| 35 | HLH | Sustained engraftment | 365 | 286 | 100 | Blood | — | — | — | 99 | 113 | Marrow |
| 36 | HIGM1 | DLI | 99 | 78 | 91 | Blood | — | — | — | 0 | 122 | T-cell |
| 37 | HLH | Death | 75 | 71 | 18 | Marrow | 70 | 19 | Blood | 18 | 71 | Marrow |
| 38 | HLH | Second TXP | 363 | 351 | 71 | Blood | 351 | 91 | Blood | 71 | 351 | Marrow |
| 39 | HLH | Second TXP | 124 | 100 | 78 | Blood | — | — | — | 78 | 100 | Marrow |
| 40 | HLH | Sustained engraftment | 365 | 238 | 100 | Blood | — | — | — | 99 | 41 | Marrow |
| 41 | HLH | Sustained engraftment | 365 | — | — | — | 216 | 100 | Blood | 95 | 81 | T-cell |
| 42 | CGD | PGF | 42 | 19 | 0 | Blood | 29 | 0 | Blood | 0 | 19 | Marrow |
| 43 | HLH | DLI | 62 | — | — | — | 50 | 46 | Blood | 17 | 68 | T-cell |
| 44 | HLH | DLI | 145 | 97 | 82 | Blood | 88 | 68 | Blood | 44 | 287 | Marrow |
| 45 | IPEX | Death | 52 | 28 | 100 | Blood | 28 | 100 | Blood | 100 | 28 | Marrow |
| 46 | CAEBV | Death | 61 | 32 | 100 | Marrow | — | — | — | 100 | 32 | Marrow |
| ID | Primary diagnosis | Sustained engraftment or failure event | Day of 1-year follow-up or failure event | Last available chimerism | Minimum recorded chimersim | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Day of unfractionated sample | Unfractionated sample, % donor | Unfractionated sample type | Day of T-cell sample | T-cell sample, % donor | T-cell sample type | % Donor | Day of sample | Sample type | ||||
| 1 | CAEBV | Sustained engraftment | 365 | — | — | — | 102 | 100 | Blood | 69 | 12 | T-cell |
| 2 | HLH | Sustained engraftment | 365 | — | — | — | 124 | 100 | Blood | 85 | 26 | T-cell |
| 3 | HLH | Death | 39 | 19 | 99 | Blood | 37 | 53 | Blood | 53 | 37 | T-cell |
| 4 | HLH | Sustained engraftment | 364 | 144 | 100 | Blood | 144 | 100 | Blood | 95 | 25 | T-cell |
| 5 | HLH | DLI | 53 | 39 | 54 | Blood | — | — | — | 4 | 18 | T-cell |
| 6 | HIGM1 | DLI | 115 | — | — | — | 98 | 97 | Blood | 92 | 76 | Marrow |
| 7 | HLH | DLI | 60 | 52 | 55 | Blood | — | — | — | 55 | 52 | Marrow |
| 8 | HLH | DLI | 73 | 70 | 24 | Blood | 70 | 37 | Blood | 2 | 170 | Marrow |
| 9 | HLH | Sustained engraftment | 365 | — | — | — | 354 | 95 | Blood | 89 | 35 | T-cell |
| 10 | HLH | DLI | 90 | 83 | 81 | Blood | 76 | 63 | Blood | 21 | 356 | Marrow |
| 11 | HLH | DLI | 74 | 73 | 28 | Blood | 73 | 82 | Blood | 5 | 307 | Marrow |
| 12 | HLH | SGF | 70 | 54 | 38 | Blood | 54 | 14 | Blood | 4 | 70 | T-cell |
| 13 | HLH | DLI | 100 | 84 | 34 | Blood | 84 | 89 | Blood | 32 | 68 | Marrow |
| 14 | HLH | Sustained engraftment | 365 | — | — | — | 245 | 95 | Blood | 93 | 175 | T-cell |
| 15 | HLH | DLI | 126 | 101 | 64 | Blood | 101 | 57 | Blood | 7 | 318 | Marrow |
| 16 | HLH | DLI | 123 | 88 | 30 | Blood | 88 | 14 | Blood | 12 | 53 | T-cell |
| 17 | HLH | Death | 253 | 248 | 100 | Blood | — | — | 100 | 24 | Marrow | |
| 18 | CAEBV | Sustained engraftment | 365 | — | — | — | 361 | 80 | Blood | 31 | 46 | T-cell |
| 19 | HLH | Death | 45 | 39 | 96 | Blood | 39 | 96 | Blood | 96 | 27 | Marrow |
| 20 | HLH | Sustained engraftment | 361 | — | — | — | 188 | 99 | Blood | 56 | 39 | T-cell |
| 21 | HLH | PGF | 42 | 41 | 0 | Blood | 41 | 0 | Blood | 0 | 27 | Marrow |
| 22 | IPEX | DLI | 138 | 105 | 15 | Blood | — | — | — | 8 | 163 | Marrow |
| 23 | HLH | Sustained engraftment | 365 | 175 | 98 | Blood | — | — | — | 97 | 371 | Marrow |
| 24 | HLH | Sustained engraftment | 363 | 333 | 90 | Blood | 333 | 97 | Blood | 90 | 27 | T-cell |
| 25 | HLH | Sustained engraftment | 365 | 275 | 100 | Blood | — | — | — | 100 | 16 | Marrow |
| 26 | CGD | SGF | 98 | — | — | — | 85 | 18 | Blood | 0 | 106 | T-cell |
| 27 | CGD | DLI | 124 | 89 | 5 | Blood | 89 | 10 | Blood | 4 | 152 | Marrow |
| 28 | HLH | Death | 96 | 84 | 100 | Blood | — | — | — | 100 | 28 | Marrow |
| 29 | HLH | Sustained engraftment | 365 | — | — | — | 362 | 100 | Blood | 100 | 21 | T-cell |
| 30 | HLH | Sustained engraftment | 365 | 312 | 100 | Blood | — | — | — | 96 | 84 | Marrow |
| 31 | HLH | Death | 246 | 243 | 99 | Blood | — | — | — | 67 | 26 | Marrow |
| 32 | CGD | Sustained engraftment | 351 | 169 | 96 | Blood | 169 | 96 | Blood | 96 | 56 | Marrow |
| 33 | HLH | Sustained engraftment | 356 | 342 | 23 | Blood | 342 | 79 | Blood | 23 | 342 | Marrow |
| 34 | CGD | Sustained engraftment | 365 | 70 | 93 | Blood | — | — | — | 93 | 70 | Marrow |
| 35 | HLH | Sustained engraftment | 365 | 286 | 100 | Blood | — | — | — | 99 | 113 | Marrow |
| 36 | HIGM1 | DLI | 99 | 78 | 91 | Blood | — | — | — | 0 | 122 | T-cell |
| 37 | HLH | Death | 75 | 71 | 18 | Marrow | 70 | 19 | Blood | 18 | 71 | Marrow |
| 38 | HLH | Second TXP | 363 | 351 | 71 | Blood | 351 | 91 | Blood | 71 | 351 | Marrow |
| 39 | HLH | Second TXP | 124 | 100 | 78 | Blood | — | — | — | 78 | 100 | Marrow |
| 40 | HLH | Sustained engraftment | 365 | 238 | 100 | Blood | — | — | — | 99 | 41 | Marrow |
| 41 | HLH | Sustained engraftment | 365 | — | — | — | 216 | 100 | Blood | 95 | 81 | T-cell |
| 42 | CGD | PGF | 42 | 19 | 0 | Blood | 29 | 0 | Blood | 0 | 19 | Marrow |
| 43 | HLH | DLI | 62 | — | — | — | 50 | 46 | Blood | 17 | 68 | T-cell |
| 44 | HLH | DLI | 145 | 97 | 82 | Blood | 88 | 68 | Blood | 44 | 287 | Marrow |
| 45 | IPEX | Death | 52 | 28 | 100 | Blood | 28 | 100 | Blood | 100 | 28 | Marrow |
| 46 | CAEBV | Death | 61 | 32 | 100 | Marrow | — | — | — | 100 | 32 | Marrow |
Acute and chronic GVHD and HLH reactivation
Eleven patients developed grade II (n = 3), grade III (n = 5), or grade IV (n = 3) acute GVHD. The day 100 and 6-month incidence of grade II to IV acute GVHD was 17.4% (95% CI, 8.1%-29.7%) and 26.1% (95% CI, 14.4%-39.4%), respectively. The corresponding incidence for grade III to IV acute GVHD was 10.9% (95% CI, 4.0%-21.9%) and 17.4% (95% CI, 8.1%-29.7%; supplemental Figure 1A), respectively. Four patients who did not receive DLI or second HCT reported acute GVHD; 2 patients reported acute GVHD before any intervention, and 5 patients after initial intervention. Twelve patients developed chronic GVHD, with its severity reported as mild (n = 10) and severe (n = 2). The 1-year cumulative incidence of chronic GVHD was 26.7% (95% CI, 14.6%-40.4%; supplemental Figure 1B). For the 5 subjects who were reported to have systemic and/or CNS reactivation, 2 HLH episodes were preceded by acute GVHD. In all the cases, the T-cell chimerism preceding HLH reactivation was more than 37% donor (Table 5).
Chimerism and GVHD data for HLH patients with reactivations
| ID | Day of HLH reactivation* | Type of HLH reactivation | Day of grade II-IV aGVHD onset* | Day of cGVHD onset* | Pre/Post HLH reactivation | Day unfractionated sample collected* | Type of unfractionated sample | Unfractionated sample, % donor | T-cell sample collected* | Type of T-cell sample | T-cell sample, % donor | Minimum recorded chimerism | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| % Donor | Day of sample | Sample type | ||||||||||||
| 8 | 139 | Systemic | 97 | 143 | Pre Post | 97 143 | Blood Blood | 10 71 | 97 143 | Blood Blood | 84 7 | 2 | 170 | Marrow |
| 15 | 314 | Systemic | No reported aGVHD | No reported cGVHD | Pre Post | 181 315 | Blood Marrow | 14 7 | 181 315 | Blood Blood | 37 14 | 7 | 318 | Marrow |
| 20 | 61 | Systemic | No reported aGVHD | 298 | Pre Post | — — | — — | — — | 39 53 | Blood Blood | 56 61 | 56 | 39 | T-cell |
| 24 | 138 328 | CNS CNS | No reported aGVHD | 155 | Pre Post Post (2nd activation) | 96 186 333 | Blood Blood Blood | 100 98 90 | 96 186 333 | Blood Blood Blood | 100 100 97 | 90 | 27 | T-cell |
| 28 | 88 | Both | 35 | No reported cGVHD | Pre Post | 56 84 | Blood Blood | 100 100 | — — | — — | — — | 100 | 28 | Marrow |
| ID | Day of HLH reactivation* | Type of HLH reactivation | Day of grade II-IV aGVHD onset* | Day of cGVHD onset* | Pre/Post HLH reactivation | Day unfractionated sample collected* | Type of unfractionated sample | Unfractionated sample, % donor | T-cell sample collected* | Type of T-cell sample | T-cell sample, % donor | Minimum recorded chimerism | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| % Donor | Day of sample | Sample type | ||||||||||||
| 8 | 139 | Systemic | 97 | 143 | Pre Post | 97 143 | Blood Blood | 10 71 | 97 143 | Blood Blood | 84 7 | 2 | 170 | Marrow |
| 15 | 314 | Systemic | No reported aGVHD | No reported cGVHD | Pre Post | 181 315 | Blood Marrow | 14 7 | 181 315 | Blood Blood | 37 14 | 7 | 318 | Marrow |
| 20 | 61 | Systemic | No reported aGVHD | 298 | Pre Post | — — | — — | — — | 39 53 | Blood Blood | 56 61 | 56 | 39 | T-cell |
| 24 | 138 328 | CNS CNS | No reported aGVHD | 155 | Pre Post Post (2nd activation) | 96 186 333 | Blood Blood Blood | 100 98 90 | 96 186 333 | Blood Blood Blood | 100 100 97 | 90 | 27 | T-cell |
| 28 | 88 | Both | 35 | No reported cGVHD | Pre Post | 56 84 | Blood Blood | 100 100 | — — | — — | — — | 100 | 28 | Marrow |
Last available chimerism data from before HLH reactivation and chimerism reported on the HLH reactivation case report form.
Day from transplant.
Regimen-related toxicity
Forty-one patients (89% of the cohort) experienced major regimen-related toxicities, including 6 who experienced fatal toxicities (13% of the cohort; 1 patient had 7 fatal toxicities reported and 1 had 3), which included fever, acute kidney injury, hemorrhage, hypotension, somnolence, capillary leak syndrome, thromboembolic event, hypoxia, and dyspnea. Cardiac and pulmonary toxicities were the most common with hypertension (grade 3: n = 30), hypotension (grade 3-5: n = 11), dyspnea (grade 3-5: n = 10), and hypoxia (grade 3-5: n = 16). Posterior reversible leukoencephalopathy was reported in 5 patients, and although 4 of these patients are dead, none of the deaths was attributed to this toxicity. Other CNS toxicities included seizures (grade 3: n = 2) and somnolence (grade 3-5: n = 4). Acute (grade 3-5: n = 7) and chronic (grade 4: n = 1) renal toxicity was reported, with 3 patients requiring dialysis. Infectious complications were common, with CMV reactivation (n = 12), EBV reactivation (n = 18), and adenovirus infection (n = 12) reported in more than 25% of all patients, and bacterial infections reported in 61% (n = 28) of patients. CNS toxicity was reported in 11% (n = 5), and only 1 patient developed veno-occlusive disease.
Immune reconstitution
Lymphocyte subsets, Ig levels, and disease-specific immune function were evaluated at baseline, day 100, and day 365 post-HCT. T cells (CD3+, CD3+4+, CD3+8+) decreased between baseline and day 100 (adjusted P < .001; Figure 3) and then increased between day 100 and 1 year to levels comparable to those recorded at baseline (adjusted P < .001 between day 100 and 1 year; P > .52 between baseline and 1 year for each). B cells (CD19+) were similar between baseline and day 100 (adjusted P = .22) and between baseline and 1 year (adjusted P = .50), but were slightly higher at 1 year than at day 100 (adjusted P = .07). NK cells (CD16+56+) were similar at each time (P > .14 for each comparison). Ig values were censored at IVIG infusion, so no formal hypothesis testing was performed for IgM, IgA, or IgG. IgG appeared to increase from baseline to day 100, but other assessments of Ig were confounded by the administration of IVIG (supplemental Table 3A). Despite normalization of NK cell number, many patients with HLH continued to demonstrate decreased or absent NK cell function even at day 365 post-HCT. Decreased or absent NK function was reported in 21 patients with HLH (72.4% of available samples) at baseline, 4 (23.5%) at day 100, and 7 (46.7%) at day 365 (supplemental Table 3B)
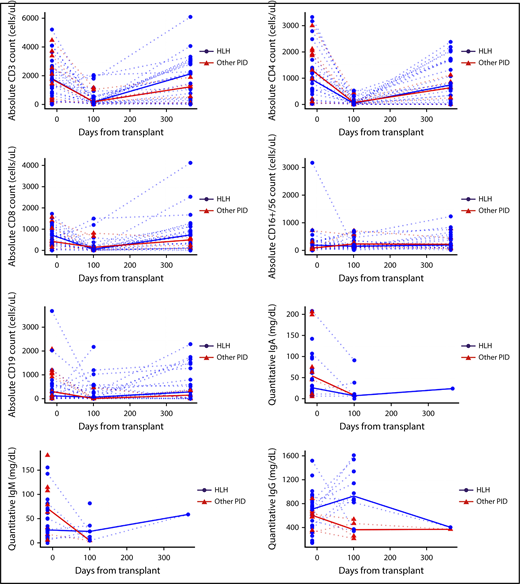
Immune reconstitution. Graphs describe absolute cell counts and quantitative Ig levels (mg/dL) with HLH (blue) and other PID patients (red), and group medians in bold lines. Quantitative IgG is censored at time of starting intravenous immune globulin (IVIG).
Immune reconstitution. Graphs describe absolute cell counts and quantitative Ig levels (mg/dL) with HLH (blue) and other PID patients (red), and group medians in bold lines. Quantitative IgG is censored at time of starting intravenous immune globulin (IVIG).
Discussion
Allogeneic HCT is required for te survival of patients with many inherited disorders of immune function. Transplantation for these patients, however, is frequently challenged by pathologic, preexisting organ dysfunction and active infections. HCT for HLH and related immune dysregulatory disorders is especially challenging because of the added risks for persistent immune activation and the potential for systemic and CNS relapse. The primary objective of this study was to evaluate the performance of the alemtuzumab (day 14), fludarabine, melphalan RIC strategy in HCT for HLH and select primary immune deficiencies in a prospective, phase 2 multicenter trial. The relatively broad confidence intervals in smaller studies make direct comparison difficult, but the 1-year survival estimate for patients with HLH (n = 34) compares with institutional RIC series, at 82.4% (90% CI, 68.4%-90.6%).9,10 The 1-year survival estimate for non-HLH diseases (n = 12) in this series was 75% (90% CI, 47.4%-89.5%), which also compares closely with published series.9,10 There were too few cases of each condition to draw disease-specific conclusions (Table 1; Figure 1), although these results demonstrate the feasibility of the intermediate RIC strategy for CAEBV and immune deficiencies.
Protection from HLH reactivation in patients with cytotoxic defects requires stable donor chimerism greater than 10% to 20%,3 and donor T cells alone may be sufficient to protect against HLH reactivation for patients with HLH as a result of defects in cytotoxicity.9 In the institutional study on which this conditioning strategy was based, survival was significantly improved compared with MAC, but patients undergoing RIC HCT in this study had extremely high rates of post-HCT mixed chimerism (65%).10 Others have also reported high rates of mixed chimerism in patients treated with alemtuzumab-based RIC regimens for HLH.9 It is therefore not surprising that a similar low rate of survival with sustained engraftment without intervention was observed in this trial, with 39% of patients (41% of HLH and 33% of non-HLH) engrafted without secondary graft failure, DLI, or second transplant and were alive at 1 year (Table 4). Despite the promising survival relative to historical MAC approaches, the risks and complications associated with unstable engraftment remain problematic with this conditioning approach (Table 2). Among the HLH cohort, recurrence was reported for 5 patients, with 1 patient reporting 2 separate reactivations. Differentiating inflammation as a result of HLH vs engraftment syndrome or immune dysregulation after HCT may be difficult, especially for early cases (<6 months post-HCT). In addition, we are now learning that some inflammasomopathies (eg, NLRC4 activation and interleukin 18) may be able to drive pathologic inflammation of donor lymphocytes.33 Diagnostic criteria for HLH have not been evaluated or validated for the post-HCT setting. However, we did track the HLH-2004 clinical criteria in patients in this cohort. Recurrence of HLH developed despite T-cell donor T-cell engraftment higher than 35% in 4 patients and whole-cell chimerism 100% in a fifth patient without T-cell data (Table 5), suggesting the hyperinflammation may have been a result of factors other than loss of protective donor T cells. Finally, it remains possible that matched donor grafts with heterozygous cytotoxic gene defects may be more sensitive to hyperinflammation during immune reconstitution and inflammatory stresses after transplant.34
The rate of grade 2 to 4 acute GVHD was higher in this study (6-month estimate of 26.2%; 26.5% for HLH) compared with the Cincinnati RIC series (8%).2 Similarly, the rate of chronic GVHD (1-year estimate, 26.7%; 32.9% for HLH) was higher in this study compared with the Cincinnati RIC series (12%; supplemental Figure 1). Another difference between results from this study and the Cincinnati RIC series is that early mortality (death before day 100) was 6 (13%) vs 0. However, the day 100 mortality rate compared favorably with historical MAC reports: 31% for HLH-94 and 50% in the Cincinnati series.1,10 Posterior reversible encephalopathy syndrome occurred in 11% of patients on this study and has been reported as a risk in patients with HLH pre-HCT, making careful control of blood pressure and neurological evaluations important components of supportive care for this group.35 Differences in outcomes in this study compared with previous HLH RIC reports may be a result of more heterogeneous patient experiences pretransplant and/or more systematic collection and reporting of adverse events and outcomes in a multicenter prospective clinical trial compared with a retrospective institutional series.
In previous series, intermediate timing alemtuzumab resulted in superior engraftment compared with more proximal or more distal timing, which was the rationale for this approach in this study.10,25 Given the variability of inflammation and lymphocyte count before transplant, a more precise pharmacokinetic approach to pretransplant alemtuzumab levels and absolute lymphocyte count may improve optimization of dosing.36 Corticosteroid GVHD prophylaxis was chosen to match the institutional trial on which this study was modeled, with the rationale that most patients entered transplant on steroids and that these agents could be protective against HLH reactivation. It is possible, however, that continued corticosteroids could also affect engraftment and susceptibility to infection. Infectious complications were common in this group: 23 participants with HLH (68%) and 8 participants with other PID (67%) reported at least 1 viral infection or reactivation of CMV, EBV, or adenovirus, for a total of 31 (67%); 19 participants with HLH (56%) and 9 with other PID (75%) reported at least 1 bacterial infection, for a total of 28 (61%). Pretransplant therapy, therapeutic agents, and poor engraftment could contribute to infection risk. Future studies could evaluate the role of alemtuzumab persistence and corticosteroid exposure on rate of infection.
This study raises the question of what the current standard of care should be for HCT for children with HLH and related primary immune deficiencies. The BMT-CTN 1204 study established rigorous, prospective data to support expectations for patients transplanted with this regimen: 1-year survival estimate, 80.4% with graft failure or additional DLI or second HCT in the majority of patients. These data provide a platform from which to design regimens that avoid excessive rates of early mortality resulting from myeloablative conditioning. Longer-term survival, however, remains suboptimal, with a major challenge of durable engraftment. This regimen could be considered a standard of care, although the high rate of suboptimal engraftment and graft failure support rapid evaluation of alternative strategies. Future studies should build on this baseline advantage of the decreased toxicity of RIC compared with MAC regiments, but improve the level and stability of donor engraftment. Potential strategies include alternative chemotherapy (eg, treosulfan),37-40 submyeloablative conditioning (eg, busulfan or thiotepa),19,41 immunotherapy (eg, CD45 antibody),42 alemtuzumab based on pharmacokinetics or absolute lymphocyte count,36 and/or targeted therapy (eg, antibody against interferon-γ).43 In addition, adaptive strategies that manipulate myelosuppression and immune suppression based on genotype, comorbidities, and donor source may further improve survival and sustained engraftment for this challenging population of children and young adults who require HCT for long-term survival.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the members of the Blood and Marrow Transplant Clinical Trials Network, the research nurses, and the patients who participated in this trial.
Support for this study was provided by grant U10HL069294 from the National Institutes of Health, National Heart, Lung, and Blood Institute and the National Institutes of Health, National Cancer Institute.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Authorship
Contribution: C.E.A., R.M., M.E., and M.A.P. conceived of the trial, reviewed data, and drafted and approved the manuscript; R.M., C.M.B., S.S., P.R., R.H., L.B., L.K., J.-A.T., K.R.S., S.-Y.P., K.S.B., J.R.A., E.O.S., and J.C. contributed to the study design, review of the data, and manuscript review; and P.D., A.R., and C.B. contributed to data analysis and manuscript review.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Carl E. Allen, 1102 Bates St, Ste 1025.22, Houston, TX 77030; e-mail: ceallen@txch.org; Michael A. Pulsipher, Division of Hematology, Oncology, and Blood & Marrow Transplantation, Children's Hospital Los Angeles, 4650 Sunset Blvd, Mailstop #54, Los Angeles, CA 90027; e-mail: mpulsipher@chla.usc.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal