

Key Points
Autosomal recessive loss-of-function mutations in G6b-B (MPIG6B) cause congenital macrothrombocytopenia with focal myelofibrosis.
G6b-B has orthologous physiological functions in human and mice regulating megakaryocyte and platelet production and function.

Abstract
Unlike primary myelofibrosis (PMF) in adults, myelofibrosis in children is rare. Congenital (inherited) forms of myelofibrosis (cMF) have been described, but the underlying genetic mechanisms remain elusive. Here we describe 4 families with autosomal recessive inherited macrothrombocytopenia with focal myelofibrosis due to germ line loss-of-function mutations in the megakaryocyte-specific immunoreceptor tyrosine-based inhibitory motif (ITIM)-containing receptor G6b-B (G6b, C6orf25, or MPIG6B). Patients presented with a mild-to-moderate bleeding diathesis, macrothrombocytopenia, anemia, leukocytosis and atypical megakaryocytes associated with a distinctive, focal, perimegakaryocytic pattern of bone marrow fibrosis. In addition to identifying the responsible gene, the description of G6b-B as the mutated protein potentially implicates aberrant G6b-B megakaryocytic signaling and activation in the pathogenesis of myelofibrosis. Targeted insertion of human G6b in mice rescued the knockout phenotype and a copy number effect of human G6b-B expression was observed. Homozygous knockin mice expressed 25% of human G6b-B and exhibited a marginal reduction in platelet count and mild alterations in platelet function; these phenotypes were more severe in heterozygous mice that expressed only 12% of human G6b-B. This study establishes G6b-B as a critical regulator of platelet homeostasis in humans and mice. In addition, the humanized G6b mouse will provide an invaluable tool for further investigating the physiological functions of human G6b-B as well as testing the efficacy of drugs targeting this receptor.
Introduction
Myelofibrosis in children is rare, and, in children, is most commonly seen in association with acute megakaryoblastic leukemia.1 Other causes include acute lymphoblastic leukemia (ALL),2 Hodgkin lymphoma,3 severe vitamin D or C deficiency,4-6 renal osteodystrophy,7 osteopetrosis, hyperparathyroidism,8 autoimmunity,9-11 and tuberculosis.12 The clinical, histologic and molecular features of pediatric myelofibrosis differ from primary myelofibrosis (PMF) in adults, which is typically due to somatic gain-of-function mutations in tyrosine kinase (TK) signaling pathways, including JAK2 V617F (50%), MPL W515K/L (5% to 10%) or CALR (35%)13,14 ; TK mutations are not typically present in pediatric myelofibrosis, particularly not in young children.1,15,16 Children with myelofibrosis have a heterogeneous phenotype with variable outcomes. Some patients have spontaneous resolution,17,18 while others have an aggressive clinical course with progressive, sometimes fatal disease cured only by hematopoietic stem cell transplantation (HSCT).15,19-21
Congenital or inherited forms of myelofibrosis (cMF) are exceedingly rare with fewer than 50 cases reported.15,18-26 Many cases occur in consanguineous families,15,19,25 suggesting that cMF can be inherited as a autosomal recessive Mendelian trait. Genetic lesions have been identified in only a small minority of cases. In some, concurrent single lineage cytopenias have been described. For example, homozygous germ line mutations in VPS45 cause neutropenia, neutrophil dysfunction, bone marrow (BM) fibrosis, progressive marrow failure and organomegaly.27,28 In some patients, fibrosis coexists with an intrinsic platelet disorder, such as gray platelet syndrome (GPS) to gain-of-function mutations in the Growth Factor Inhibitor 1B (GFI1B) gene29 or biallelic mutations in neurobeachin-like 2 (NBEAL2).30-32 These phenotypes are quite distinct from most patients with cMF.
Here we describe the clinical phenotype, distinctive histological features and biallelic loss-of-function mutations in the megakaryocyte-specific immunoreceptor tyrosine-based inhibitor motif (ITIM)-containing receptor G6b-B (C6orf25 or MPIG6B, referred to as G6b) in four families with a clinically distinctive form of cMF associated with macrothrombocytopenia and focal myelofibrosis (cMTFM). These phenotypes closely resemble those reported in G6b knockout (KO) and loss-of-function mice (Geer et al).33,34 A humanized G6b transgenic knockin (KI) mouse model establishes that human and mouse G6b-B perform orthologous functions. Mice homozygous for human G6b (G6bhu/hu) exhibit a mild platelet disorder attributed to reduced human G6b-B expression. Reduced G6b-B expression in G6bhu/hu and heterozygous (G6bhu/−) mice also revealed novel regulatory roles of G6b-B with respect to immunoreceptor tyrosine-based activation motif (ITAM) and hemi-ITAM receptor signaling.
Materials and methods
Patients
Families 1-3 consented to the Pediatric Myelodysplastic Syndrome and Bone Marrow Failure Registry (Boston Children’s Hospital, Protocol #10-02-0057). Family 4 was consented to the Biomedical Research Centres/Units Inherited Diseases Genetic Evaluation-Bleeding and Platelet Disorders (BRIDGE-BPD) study (UK REC10/H0304/6). All studies were conducted according to the Declaration of Helsinki.
Hematological characterization and sample preparation
Complete blood counts (CBC) and BM aspirates and biopsies were obtained on all clinically affected individuals; CBCs were obtained on other family members. Whole blood or BM mononuclear cell DNA was extracted using the Qiagen Puregene Blood Kit (Qiagen, Germantown, MD). BM aspirate and biopsy histology were prepared using standard clinical procedures. Images were obtained with an Olympus BX43 microscope and DP25 digital camera and acquired with the Olympus CellSens Entry Imaging Software.
Flow cytometry, immunohistochemistry, and immunoblotting
Flow cytometry was performed on an BD Accuri C6 flow cytometer using methods and antibodies described in the supplemental Methods (available on the Blood Web site). Immunohistochemical (IHC) staining for CD61 was done on the Bouin-fixed, paraffin-embedded tissue sections using the Ventana Discovery XT automated platform. A similar, custom automated IHC method was developed using a monoclonal antibody directed against the N-terminal ectodomain of G6b-B.33 western blotting and immunoprecipitations were performed as previously described.35
Linkage and sequencing analysis
Affymetrix 6.0 SNP genotyping (QuintilesIMS, Research Triangle Park, NC) was performed on members of the index family (family 1) and analyzed using a custom platform (Variant Explorer, K.S.-A. and K.M., unpublished). Whole exome sequencing (WES) was performed in patients and immediate family members of family 1 using the Illumina TrueSeq enrichment protocol. Families 2 and 3 were sequenced using the Agilent SureSelect exome capture enrichment protocol. Sequencing was performed on an Illumina Hisequation 2000/2500 with 100 bp paired-end sequencing with an average depth of 40-200x (Macrogen, Rockville, MD). FASTq sequences were processed and analyzed using the WuXi/NextCode platform (Cambridge, MA). For individual 4-II-3, DNA library preparation, sequencing, variant calling and variant filtering were performed as described previously.36 Confirmatory Sanger sequencing was carried out on all patients and available family members as described in the supplemental Methods.
Mouse models
Humanized G6b KI mice were generated as outlined in supplemental Figure 1 and supplementary material (Taconic Biosciences, Cologne, Germany). G6b constitutive KO (G6b−/−) mouse models were generated as previously described.33 All mice were on a C57BL/6 background and procedures were undertaken with UK Home Office approval, in accordance with the Animals (Scientific Procedures) Act of 1986. Detailed descriptions of all other methods can be found in supplemental Methods.
Results
Clinical and pathological characteristics
We studied four consanguineous Arab families presenting with macrothrombocytopenia, anemia and myelofibrosis. Family 1, from the United Arab Emirates (UAE), had three affected male siblings born to first cousin parents. Affected individuals (Figure 1A; Table 1, 1-III-1, 1-III-5, and 1-III-6) had macrothrombocytopenia with variably reduced platelet granularity, and microcytic anemia. BM studies revealed clusters of atypical megakaryocytes with reticulin fibrosis that was most pronounced in proximity to the megakaryocytes. Affected children had mild to moderate bleeding symptoms and required frequent platelet and occasional red cell transfusions; each affecte individual shared a homozygous human leukocyte antigen (HLA) haplotype and had congenital adrenal hyperplasia (CAH) due to 21-hydroxylase (CYP21A2) deficiency. The children had three apparently unaffected cousins born to siblings of their parents. One female cousin had CAH and shared the same homozygous HLA type as her affected cousins (Figure 1A; Table 1, 1-III-9,). She was not thrombocytopenic and had only rare large platelets on her peripheral blood smear (Figure 2C). While being evaluated as a transplant donor for her cousins, she underwent multiple BM studies, the first of which showed several lymphoid aggregates with associated reticulin, but normal megakaryocytes. Follow-up BM studies (Figure 2F,I) were normal. Given the lack of a sibling, unrelated or cord blood donor, persistent transfusion dependency and lack of response to immunomodulatory agents, patients 1-III-5 and 1-III-6 underwent successful matched related HSCTs with 1-III-9 serving as the donor. Both patients engrafted and are doing well following HSCT. The proband in this family, 1-III-1, was transplanted using a haploidentical approach from his mother, and eventually succumbed to transplant-related complications.15
![Figure 1. cMTFM is due to mutations in G6b-B (G6b). (A) Family pedigrees. Black arrows point to the probands in each family. Double line indicates a consanguineous relationship. Ages are provided at the time of the initial evaluation of the youngest affected individual in the family. (B) Ideograms showing cMTFM linkage region, the G6b (MPIG6B) locus and patient mutations. (C) Expression of p.Gly157Arg mutant in DT40 cells leads to protein instability and (D-E) reduced expression on the surface of the cell (mean ± standard error of the mean [SEM]; n = 2). pcDNA3 vector, blue; G6b-B WT, green; G6b-B p.Gly157Arg, red. Representative blots and histograms of 2 independent experiments. MFI, median fluorescence intensity.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/132/13/10.1182_blood-2017-08-802769/4/m_blood802769f1.png?Expires=1769093884&Signature=jcB10JOyQOje3XkvUCUPWA1BDMGgm0adgNkjs3ORFNygqGND62nWUWCu8FHYNB5m7xmrxq1g1CaxuyE8njN1BM5O7KfqBHhhFxEf1U2CvsKYu7nLpZqSmNZu3rq0GRZPU85UVWCKxYUlk69RGFVBskiMMFFnDrioFqFsoYVz5Jia~BoaUCI70LPaFYUoZ96MPRedpELpEgAOaqJ1xTXDO5UjWJbsBD53kTO~cj0tCXeT3oGqbXBMdMMGR3Gf0OIv7JAzdR7nNxZndZbhdt5wz-ybtgmExW1vE4-vTazOdNH7Oo5yZU62kt-7OCSv8LH6IcXvqp6pjXS0qhgbkc88RQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
cMTFM is due to mutations in G6b-B (G6b). (A) Family pedigrees. Black arrows point to the probands in each family. Double line indicates a consanguineous relationship. Ages are provided at the time of the initial evaluation of the youngest affected individual in the family. (B) Ideograms showing cMTFM linkage region, the G6b (MPIG6B) locus and patient mutations. (C) Expression of p.Gly157Arg mutant in DT40 cells leads to protein instability and (D-E) reduced expression on the surface of the cell (mean ± standard error of the mean [SEM]; n = 2). pcDNA3 vector, blue; G6b-B WT, green; G6b-B p.Gly157Arg, red. Representative blots and histograms of 2 independent experiments. MFI, median fluorescence intensity.
cMTFM is due to mutations in G6b-B (G6b). (A) Family pedigrees. Black arrows point to the probands in each family. Double line indicates a consanguineous relationship. Ages are provided at the time of the initial evaluation of the youngest affected individual in the family. (B) Ideograms showing cMTFM linkage region, the G6b (MPIG6B) locus and patient mutations. (C) Expression of p.Gly157Arg mutant in DT40 cells leads to protein instability and (D-E) reduced expression on the surface of the cell (mean ± standard error of the mean [SEM]; n = 2). pcDNA3 vector, blue; G6b-B WT, green; G6b-B p.Gly157Arg, red. Representative blots and histograms of 2 independent experiments. MFI, median fluorescence intensity.
Clinical and laboratory characteristics, treatments, and outcomes of patients with congenital myelofibrosis
| ID | Age/Sex | WBC ×109/L | HGB (g/dL) | PLT ×109/L | MCV (fL) | MPV (fL) | PB smear morphology | Other clinical features | JAK2 | MPL | CALR | Therapy and response | HLA | G6B genotype | Outcome | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A | B | C | DRB1 | DQB1 | |||||||||||||||
| 1-III-1 | 5 mo/M | NA | 6.0 | 20 | 66.0 | NA | Microcytosis, anisopoikilocytosis, target cells, ovalocytes, large platelets | CAH | Neg | ND | ND | No pre-HSCT therapy | 11:01 | 41:01 | 07:01 | 08:04 | 03:01 | c.61_61+1dup/ c.61_61+1dup | Deceased from HSCT complications, graft failure after 2nd HSCT, GI bleed, ARDS |
| S/P 2 haploidentical HSCT from mother | 11:01 | 41:01 | 07:01 | 08:04 | 03:01 | ||||||||||||||
| 1-III-5 | Birth/M | NA | 6.9 | 16 | NA | NA | Anisopoikilocytosis, target cells, basophilic stippling, large platelets | CAH | Neg | Neg | Neg | Steroids, transient partial response but not sustained. Platelets declined with taper. | 11:01 | 41:01 | 07:01 | 08:04 | 03:01 | c.61_61+1dup/ c.61_61+1dup | 6 y post-HSCT, engrafted, maturing trilineage hematopoiesis with no evidence of fibrosis. |
| MRD HSCT from 1-III-9 | 11:01 | 41:01 | 07:01 | 08:04 | 03:01 | ||||||||||||||
| 1-III-6 | 6 mo/M | 13.79 | 8.7 | 19 | 68.0 | NA | Microcytosis, anisopoikilocytosis, target cells, basophilic stippling, tear drops, frequent large platelets | CAH | Neg | Neg | Neg | No pre-HSCT therapy. | 11:01 | 41:01 | 07:01 | 08:04 | 03:01 | c.61_61+1dup/ c.61_61+1dup | 4 y post-HSCT. Slow platelet and RBC engraftment. Maturing trilineage hematopoiesis and no evidence of fibrosis, but platelets remain decreased, ∼119 × 109/L-142 × 109/L |
| MRD HSCT from 1-III-9 | 11:01 | 41:01 | 07:01 | 08:04 | 03:01 | ||||||||||||||
| 1-III-9 | 2 y/F | 8.39 | 11.9 | 468 | 72.0 | 8.2 | Microcytosis | CAH, mild iron deficiency | ND | ND | ND | None | 11:01 | 41:01 | 07:01 | 08:04 | 03:01 | c.61_61+1dup/ c.61_61+1dup | Clinically unaffected, rare giant platelets |
| 11:01 | 41:01 | 07:01 | 08:04 | 03:01 | |||||||||||||||
| 2-II-5 | 7 y/M | 5.43 | 11.7 | 107 | 80.5 | NA | Anisopoikilocytosis, microcytosis, ovalocytes, burr cells, helmet cells, acanthocytes, schistocytes, frequent large platelets | None | ND | ND | ND | None | 23:01 | 50:01 | 06:02 | 04:06 | 04:02 | c.147insT | Observation, stable disease |
| 23:01 | 50:01 | 06:02 | 04:06 | 04:02 | p.Ala52GlyfsX128 | ||||||||||||||
| 2-II-6 | 6 mo/M | NA | 5.4 | 10 | 65.0 | 12-24 | Anisopoikilocytosis, microcytosis, elliptocytes, schistocytes, frequent large platelets | None | Neg | Neg | Neg | IVIG ×1 | 23:01 | 50:01 | 06:02 | 04:06 | 04:02 | c.147insT | Observation, stable disease |
| Steroids ×1 with transient responses | 23:01 | 50:01 | 06:02 | 04:06 | 04:02 | p.Ala52GlyfsX128 | |||||||||||||
| 3-II-4 | 17 mo/F | 19.2 | 5.1 | 81 | 71.3 | NA | Anisopoikilocytosis, microcytosis, target cells, ovalocytes, burr cells, tear drop cells, schistocytes, frequent large platelets | CAH, asplenia | Neg | Neg | Neg | None | 11:01 | 41:01 | 07:01 | 08:04 | 03:01 | c.61_61+1dup/ c.61_61+1dup | Observation, stable disease |
| 11:01 | 41:01 | 07:01 | 08:04 | 03:01 | |||||||||||||||
| 4-II-2 | 18 mo/M | NA | 5.4 | 96 | NA | NA | Anisopoikilocytosis, target cells, schistocytes, leptocytes, dacryocytes, Howell-Jolly bodies | Cerebral cavernoma | ND | Neg | ND | IVIG and steroid, no response | ND | ND | ND | ND | ND | c.469G>A | Observation, stable thrombocytopenia, supportive care with platelets |
| p.Gly157Arg | |||||||||||||||||||
| 4-II-3 | 1 y/F | 17 | 12.5 | 97 | NA | NA | Anisopoikilocytosis, target cells, schistocytes, leptocytes, dacryocytes, Howell-Jolly bodies | None | ND | ND | ND | None | ND | ND | ND | ND | ND | c.469G>A | Observation, stable mild/moderate thrombocytopenia |
| p.Gly157Arg | |||||||||||||||||||
| ID | Age/Sex | WBC ×109/L | HGB (g/dL) | PLT ×109/L | MCV (fL) | MPV (fL) | PB smear morphology | Other clinical features | JAK2 | MPL | CALR | Therapy and response | HLA | G6B genotype | Outcome | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A | B | C | DRB1 | DQB1 | |||||||||||||||
| 1-III-1 | 5 mo/M | NA | 6.0 | 20 | 66.0 | NA | Microcytosis, anisopoikilocytosis, target cells, ovalocytes, large platelets | CAH | Neg | ND | ND | No pre-HSCT therapy | 11:01 | 41:01 | 07:01 | 08:04 | 03:01 | c.61_61+1dup/ c.61_61+1dup | Deceased from HSCT complications, graft failure after 2nd HSCT, GI bleed, ARDS |
| S/P 2 haploidentical HSCT from mother | 11:01 | 41:01 | 07:01 | 08:04 | 03:01 | ||||||||||||||
| 1-III-5 | Birth/M | NA | 6.9 | 16 | NA | NA | Anisopoikilocytosis, target cells, basophilic stippling, large platelets | CAH | Neg | Neg | Neg | Steroids, transient partial response but not sustained. Platelets declined with taper. | 11:01 | 41:01 | 07:01 | 08:04 | 03:01 | c.61_61+1dup/ c.61_61+1dup | 6 y post-HSCT, engrafted, maturing trilineage hematopoiesis with no evidence of fibrosis. |
| MRD HSCT from 1-III-9 | 11:01 | 41:01 | 07:01 | 08:04 | 03:01 | ||||||||||||||
| 1-III-6 | 6 mo/M | 13.79 | 8.7 | 19 | 68.0 | NA | Microcytosis, anisopoikilocytosis, target cells, basophilic stippling, tear drops, frequent large platelets | CAH | Neg | Neg | Neg | No pre-HSCT therapy. | 11:01 | 41:01 | 07:01 | 08:04 | 03:01 | c.61_61+1dup/ c.61_61+1dup | 4 y post-HSCT. Slow platelet and RBC engraftment. Maturing trilineage hematopoiesis and no evidence of fibrosis, but platelets remain decreased, ∼119 × 109/L-142 × 109/L |
| MRD HSCT from 1-III-9 | 11:01 | 41:01 | 07:01 | 08:04 | 03:01 | ||||||||||||||
| 1-III-9 | 2 y/F | 8.39 | 11.9 | 468 | 72.0 | 8.2 | Microcytosis | CAH, mild iron deficiency | ND | ND | ND | None | 11:01 | 41:01 | 07:01 | 08:04 | 03:01 | c.61_61+1dup/ c.61_61+1dup | Clinically unaffected, rare giant platelets |
| 11:01 | 41:01 | 07:01 | 08:04 | 03:01 | |||||||||||||||
| 2-II-5 | 7 y/M | 5.43 | 11.7 | 107 | 80.5 | NA | Anisopoikilocytosis, microcytosis, ovalocytes, burr cells, helmet cells, acanthocytes, schistocytes, frequent large platelets | None | ND | ND | ND | None | 23:01 | 50:01 | 06:02 | 04:06 | 04:02 | c.147insT | Observation, stable disease |
| 23:01 | 50:01 | 06:02 | 04:06 | 04:02 | p.Ala52GlyfsX128 | ||||||||||||||
| 2-II-6 | 6 mo/M | NA | 5.4 | 10 | 65.0 | 12-24 | Anisopoikilocytosis, microcytosis, elliptocytes, schistocytes, frequent large platelets | None | Neg | Neg | Neg | IVIG ×1 | 23:01 | 50:01 | 06:02 | 04:06 | 04:02 | c.147insT | Observation, stable disease |
| Steroids ×1 with transient responses | 23:01 | 50:01 | 06:02 | 04:06 | 04:02 | p.Ala52GlyfsX128 | |||||||||||||
| 3-II-4 | 17 mo/F | 19.2 | 5.1 | 81 | 71.3 | NA | Anisopoikilocytosis, microcytosis, target cells, ovalocytes, burr cells, tear drop cells, schistocytes, frequent large platelets | CAH, asplenia | Neg | Neg | Neg | None | 11:01 | 41:01 | 07:01 | 08:04 | 03:01 | c.61_61+1dup/ c.61_61+1dup | Observation, stable disease |
| 11:01 | 41:01 | 07:01 | 08:04 | 03:01 | |||||||||||||||
| 4-II-2 | 18 mo/M | NA | 5.4 | 96 | NA | NA | Anisopoikilocytosis, target cells, schistocytes, leptocytes, dacryocytes, Howell-Jolly bodies | Cerebral cavernoma | ND | Neg | ND | IVIG and steroid, no response | ND | ND | ND | ND | ND | c.469G>A | Observation, stable thrombocytopenia, supportive care with platelets |
| p.Gly157Arg | |||||||||||||||||||
| 4-II-3 | 1 y/F | 17 | 12.5 | 97 | NA | NA | Anisopoikilocytosis, target cells, schistocytes, leptocytes, dacryocytes, Howell-Jolly bodies | None | ND | ND | ND | None | ND | ND | ND | ND | ND | c.469G>A | Observation, stable mild/moderate thrombocytopenia |
| p.Gly157Arg | |||||||||||||||||||
ARDS, acute respiratory distress syndrome; CAH, congenital adrenal hyperplasia; F, female; GI, gastrointestinal; HGB, hemoglobin; HSCT, hematopoietic stem cell transplant; IVIG, IV immunoglobulin; M, male; MCV, mean corpuscular volume; MPV, mean platelet volume; MRD, minimal residual disease; NA, not available; ND, not done; Neg, negative; PB, peripheral blood; PLT, platelet; RBC, red blood cell; S/P, status post; WBC, white blood cell.
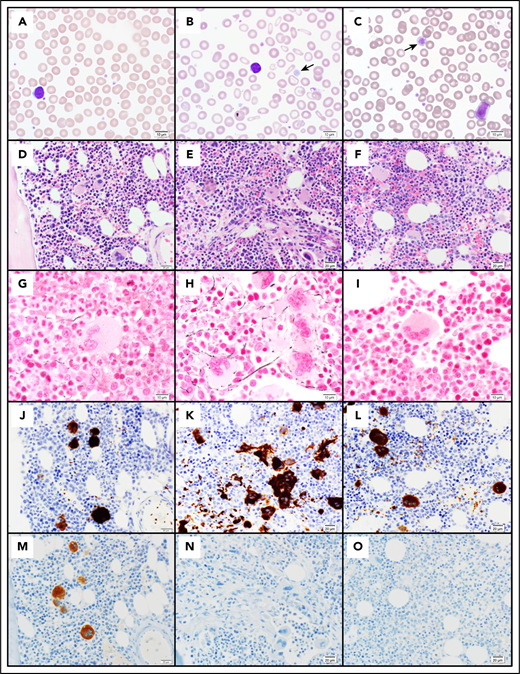
Pathology of cMTFM. Wright-Giemsa–stained peripheral blood smears from (A) a normal control, (B) patient 3-II-4 and (C) the clinically unaffected, genotypically G6b-mutated individual 1-III-9 (original magnification ×1000). Note the large, hypogranular platelets (arrow) and RBC anisocytosis in 3-II-4 and the rare giant platelet in 1-III-9 (arrow). Serial histologic sections of a bone marrow biopsies from (D,G,J,M) a normal control individual, (E,H,K,N) 3-II-4, and (F,I,L,O) 1-III-9 stained with (D-F) H&E (original magnification ×400), (G-I) Reticulin (original magnification ×600), (J-L) the megakaryocytic marker CD61 (original magnification ×400), and (M-O) G6b-B (original magnification ×400). Atypical megakaryocytes present in stellate clusters associated with increased reticulin staining are characteristic of cMTMF. Staining for G6b-B is entirely negative in the megakaryocytes and platelets from 3-II-4 and 1-III-9.
Pathology of cMTFM. Wright-Giemsa–stained peripheral blood smears from (A) a normal control, (B) patient 3-II-4 and (C) the clinically unaffected, genotypically G6b-mutated individual 1-III-9 (original magnification ×1000). Note the large, hypogranular platelets (arrow) and RBC anisocytosis in 3-II-4 and the rare giant platelet in 1-III-9 (arrow). Serial histologic sections of a bone marrow biopsies from (D,G,J,M) a normal control individual, (E,H,K,N) 3-II-4, and (F,I,L,O) 1-III-9 stained with (D-F) H&E (original magnification ×400), (G-I) Reticulin (original magnification ×600), (J-L) the megakaryocytic marker CD61 (original magnification ×400), and (M-O) G6b-B (original magnification ×400). Atypical megakaryocytes present in stellate clusters associated with increased reticulin staining are characteristic of cMTMF. Staining for G6b-B is entirely negative in the megakaryocytes and platelets from 3-II-4 and 1-III-9.
Family 2 (Figure 1) is from Saudi Arabia. The proband (2-II-6), whose parents are first cousins, had transient thrombocytopenia at birth. Thrombocytopenia recurred at 6-months of life and was associated with severe microcytic anemia unresponsive to iron therapy. BM studies performed elsewhere at age 2-years were reported to be normal. Severe thrombocytopenia and anemia persisted despite several courses of intravenous immunoglobulin and steroids. A BM at age 3-years showed a hypercellular marrow with increased lymphocytes, and clustering of atypical megakaryocytes with focal reticulin fibrosis. The patient’s older brother (2-II-5) was found to be an HLA identical match, but was ineligible as a donor due to moderate macrothrombocytopenia. BM studies are not available for this individual, though he is presumed to be clinically affected. Given the lack of a suitable donor and their mild phenotype requiring only occasional platelet transfusions, both children continue to be monitored.
The proband in family 3 (Figure 1A, 3-II-4) is the daughter of first cousin consanguineous parents from the UAE. She presented at 15-months of age with leukocytosis, severe microcytic anemia and macrothrombocytopenia. Her clinical features were strikingly similar to the affected children in family 1. Her BM showed a mildly hypercellular marrow for her age with a relative myeloid hyperplasia and clusters of atypical megakaryocytes with associated reticulin fibrosis. Although family 1 and family 3 are not known to be related, 3-II-4 also has CAH and shares the same homozygous HLA type and CYP21A2 mutation as family 1. She is currently being followed without therapeutic intervention. JAK2, MPL, and CALR mutation analysis was negative in patients from families 1-3 (Table 1).
The fourth family included an affected male and female sibling pair with macrothrombocytopenia, increased BM megakaryocytes, and grade II BM myelofibrosis (Figure 1A, 4-II-2, 4-II-3). These children and an unaffected brother were born to first cousins of Arab descent. The affected boy (4-II-2) had a more severe phenotype, including anemia with Howell-Jolly bodies, elevated fetal hemoglobin, dyserythropoiesis and dysmegakaryopoiesis. Platelet surface expression of GPIbα, αIIb, α2 and GPIV were all unaltered, and there was a minor reduction in aggregation to ADP (data not shown).
A detailed description of each case is included in the supplemental Data: clinical case descriptions.
Linkage, mutational and protein expression analyses
We performed Affymetrix SNP 6.0 genotyping on peripheral blood DNA from 10 individuals from family 1 and assessed shared regions of homozygosity by descent in 1-III-5 and 1-III-6; 1-III-9 was regarded as having an ambiguous phenotype. The major histocompatibility locus (MHC) on chromosome 6p, which, although not statistically significant, showed the strongest linkage (LOD = 2.01; Figure 1B). We focused on this region, because affected individuals in family 1 and 3 had an identical homozygous HLA type and congenital adrenal hyperplasia (CAH) due to the identical CYP21A2 mutation (Table 1). Furthermore, both affected individuals in family 2 had a different, but homozygous, HLA haplotype; the CYP21A2 and HLA loci are located at 6p21.33 and 6p21.32-6p22.1, respectively (Figure 1B).
We performed WES on nuclear families 1-3, and on individual 4-II-3 and interrogated the results for genes in which unambiguously affected members of each family carried a homozygous rare variant (allele frequency <0.005) within the candidate disease interval predicted to be functionally deleterious. The only gene meeting these criteria was the megakaryocyte and platelet-specific receptor G6b (MPIG6B, C6orf25, referred to as G6b-B or G6b).
G6b-B is a transmembrane receptor expressed on the surface of platelets and megakaryocytes.33,37-39 G6b-B has one extracellular immunoglobulin-like domain, a single transmembrane domain and an immunoreceptor tyrosine-based inhibitory motif (ITIM) and immunoreceptor tyrosine-based switch motif (ITSM) in its cytoplasmic tail. Tyrosine phosphorylation of the ITIM and ITSM provides a docking site for the SRC homology 2 (SH2) domain-containing tyrosine phosphatases SHP1 (PTPN6) and SHP2 (PTPN11), which mediate downstream signaling.40,41 Murine KO and loss-of-function models of G6b result in a phenotype very similar to the patients, including macrothrombocytopenia, abnormal platelet function and the perimegakaryocytic pattern of bone marrow fibrosis present in patients (Geer et al).33,34 BM-derived mouse G6b-B-deficient megakaryocytes develop and proliferate normally in vitro, but failed to form highly branched proplatelets, suggesting that G6b-B regulates proplatelet formation, correlating with reduced platelet production.33
Families 1 and 3 shared the same variant, a two base pair duplication bridging a splice donor site, NM_138272.2 c.61_61+1dup (p.?), and family 2 had a unique homozygous frameshift mutation, c.149dup (p.Ala52GlyfsX128; Figure 1B). These variants segregated with the phenotype in families 2 and 3; however, in family 1, individual 1-III-9, who had served as the HSCT donor for her cousins, genotyped as a homozygous mutant, despite having no overt clinical features of the disease other than rare giant platelets (Figure 2C). In family 4, we found a novel apparently homozygous missense variant c.469G>A (p.Gly157Arg; Figure 1B). The variant was also homozygous in the affected brother.
Residues 143-163 encode the predicted transmembrane domain of G6b-B, and substituting a glycine with arginine would likely destabilize insertion of the receptor into the membrane. Expression of the p.Gly157Arg mutant protein was tested using transiently transfected DT40 cells. Introduction of the mutation caused a significant reduction in total G6b-B protein expression (Figure 1C), and surface expression was reduced by 84% by flow cytometry (Figure 1D-E). A comparable reduction in expression was also observed in transiently transfected Chinese hamster ovary (CHO) cell line (data not shown). These data suggest that this rare missense variant is functionally significant.
To further validate G6b variants as disease-causing, we evaluated G6b-B expression in BM biopsy specimens from affected patients and control samples by IHC using a monoclonal antibody specific to the ectodomain. G6b-B was strongly and selectively expressed in megakaryocytes and platelets of control BM samples, but completely absent in megakaryocytes and platelets of the genotypically affected individuals from families 1-3 (Figure 2J-O). BM biopsy samples were not available for patient 4-II-2 or 4-II-3 with the p.Gly157Arg mutation. Thus, in combination with the complementary phenotype observed in G6b−/− mice,33 the thrombocytopenia and BM myelofibrosis found in these patients is almost certainly due to reduced or absent expression of G6b-B.
Expression of human G6b-B in G6bhu/hu mice
To confirm that human and mouse G6b-B are orthologous, we generated a humanized G6b mouse model in which mouse G6b was replaced with its human counterpart (supplemental Figure 1; supplemental Methods). Mice were born at expected Mendelian frequencies from heterozygous (G6b+/hu) breeding pairs (supplemental Table 1), and G6bhu/hu mice were healthy, fertile and survived >25 weeks without any overt developmental or growth anomalies. Expression of human and mouse G6b-B was verified by flow cytometry (supplemental Figure 2) and western blotting (Figure 3A). Expression of the human G6b-A splice isoform, which lacks the ITIM and ITSM, was also demonstrated using an isoform-specific antibody (Figure 3Ai,iv). Both human isoforms were only expressed at 25% of normal human platelet levels. The ratio of glycosylated (32 kDa) to unglycosylated (28 kDa) human G6b-B and -A was reduced from 3:2 in human platelets to 2:3 in G6bhu/hu platelets (Figure 3B).
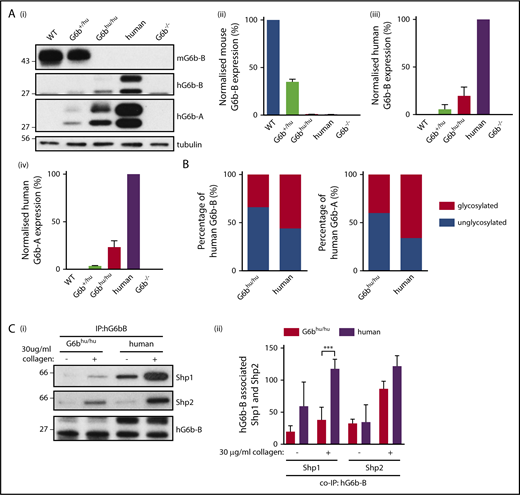
Expression of G6b isoforms in humanized mouse model. (A) WT, G6b+/hu, G6bhu/hu, human and G6b−/− washed platelet lysates (4 × 108/mL), were resolved by SDS-PAGE and custom antibodies used to detect mouse G6b-B (mG6b-B), human G6b-B (hG6b-B ), and hG6b-A. Expression was quantified and normalized to (i,ii) WT or (iii,iv) human lysates, to show relevant expression levels in the mouse models. (B) Quantification of glycosylated and unglycosylated human G6b-A and -B, data normalized to show percentage of total expression. (C) hG6b-B was immunoprecipitated (IP) from basal and collagen-activated G6bhu/hu and human washed platelet lysates (4 × 108/mL). Co-IP of Shp1 and Shp2 was investigated by (i) SDS-PAGE before (ii) quantification and normalization to hG6b-B levels. Quantification was completed using Licor Odyssey system. All data represented as mean ± SEM (n = 3-4). ***P < .001.
Expression of G6b isoforms in humanized mouse model. (A) WT, G6b+/hu, G6bhu/hu, human and G6b−/− washed platelet lysates (4 × 108/mL), were resolved by SDS-PAGE and custom antibodies used to detect mouse G6b-B (mG6b-B), human G6b-B (hG6b-B ), and hG6b-A. Expression was quantified and normalized to (i,ii) WT or (iii,iv) human lysates, to show relevant expression levels in the mouse models. (B) Quantification of glycosylated and unglycosylated human G6b-A and -B, data normalized to show percentage of total expression. (C) hG6b-B was immunoprecipitated (IP) from basal and collagen-activated G6bhu/hu and human washed platelet lysates (4 × 108/mL). Co-IP of Shp1 and Shp2 was investigated by (i) SDS-PAGE before (ii) quantification and normalization to hG6b-B levels. Quantification was completed using Licor Odyssey system. All data represented as mean ± SEM (n = 3-4). ***P < .001.
The ability of human G6b-B to bind mouse Shp1 and Shp2 was also investigated in G6bhu/hu platelets (Figure 3C). Similar amounts of human and mouse Shp1 and Shp2 co-immunoprecipitated (co-IP’d) with human G6b-B from human and G6bhu/hu platelets under resting conditions (Figure 3Cii). However, less Shp1 co-IP’d with human G6b-B from G6bhu/hu platelets compared with human platelets following collagen-stimulation, most likely reflecting differences in expression levels of Shp1 and Shp2 in human and mouse platelets (supplemental Figure 3).42,43 In contrast, comparable amounts of Shp2 co-IP’d with human G6b-B from collagen-stimulated human and G6bhu/hu platelets.
Human G6b-B functionally replaces mouse G6b-B
Reduced human G6b-B expression resulted in an 21% reduction in platelet count and a marginal increase in platelet volume in G6bhu/hu mice (Table 2; supplemental Figure 4). In contrast, complete ablation of G6b resulted in a >80% reduction in platelet count and >30% increase in platelet volume (Table 2), demonstrating that human G6b-B rescues the KO phenotype. Altered blood cell counts in G6b KO mice were independent of age (supplemental Figure 5). Histological sections of spleens and femurs also demonstrated that expression of human G6b rescues the perimegakaryocytic pattern of bone marrow fibrosis previously identified in G6b KO mice (supplemental Figure 6).33 All other hematological parameters were normal in G6bhu/hu mice (Table 2). Blood counts were normal in G6b+/hu and G6b+/− mice (Table 2).33 Platelet surface levels of GPVI, CLEC-2, α2, αIIb and GPIbα were normal in platelets from G6bhu/hu mice (supplemental Figure 7).
Peripheral blood counts of indicated mouse genotypes
| Hematological parameters | WT, n = 36 | G6b+/hu, n = 37 | G6bhu/hu, n = 37 | G6bhu/−, n = 31 | G6b−/−, n = 37 | λ | P |
|---|---|---|---|---|---|---|---|
| Platelets, 109/L | 1118 ± 209 | 1127 ± 219 | 884 ± 214*** | 795 ± 187*** | 206 ± 86*** | 0.2 | 2 × 10−72 |
| Mean platelet volume, fL | 5.8 ± 0.2 | 5.8 ± 0.2 | 6.1 ± 0.5*** | 6.2 ± 0.4*** | 7.8 ± 0.4*** | −2 | 2 × 10−65 |
| Plateletcrit, % | 0.4 ± 0.1 | 0.4 ± 0.1 | 0.3 ± 0.1 | 0.3 ± 0.1 | 0.1 ± 0 | — | — |
| Red blood cells, 1012/L | 11.1 ± 0.7 | 10.5 ± 0.7 | 10.7 ± 0.8 | 10.4 ± 0.6 | 10.0 ± 0.6 | — | — |
| Hematocrit, % | 33.6 ± 2.4 | 32.7 ± 1.8 | 33.1 ± 2.5 | 34.1 ± 1.9 | 31.8 ± 1.7 | — | — |
| White blood cells, 109/L | 7.5 ± 3.6 | 8.6 ± 2.3 | 7.4 ± 2.6 | 10.1 ± 6.1 | 13.1 ± 6.8*** | 0.5 | 8 × 10−7 |
| Lymphocytes, 109/L | 6.2 ± 2.7 | 7.4 ± 2.0 | 6.9 ± 2.2 | 7.8 ± 3.7 | 11.8 ± 4.9*** | 0.5 | 3 × 10−11 |
| Monocytes, 109/L | 0.5 ± 0.4 | 0.3 ± 0.2 | 0.5 ± 0.2 | 0.3 ± 0.3 | 0.5 ± 0.4 | — | — |
| Neutrophils, 109/L | 0.8 ± 0.4 | 0.9 ± 0.4 | 0.7 ± 0.3 | 1.2 ± 0.8 | 1.7 ± 1.1 | — | — |
| Eosinophils, 109/L | 0.1 ± 0.2 | 0 ± 0 | 0 ± 0 | 0 ± 0.1 | 0 ± 0 | — | — |
| Basophils, 109/L | 0.1 ± 0.3 | 0 ± 0 | 0.1 ± 0.2 | 0 ± 0.1 | 0.1 ± 0.1 | — | — |
| Hematological parameters | WT, n = 36 | G6b+/hu, n = 37 | G6bhu/hu, n = 37 | G6bhu/−, n = 31 | G6b−/−, n = 37 | λ | P |
|---|---|---|---|---|---|---|---|
| Platelets, 109/L | 1118 ± 209 | 1127 ± 219 | 884 ± 214*** | 795 ± 187*** | 206 ± 86*** | 0.2 | 2 × 10−72 |
| Mean platelet volume, fL | 5.8 ± 0.2 | 5.8 ± 0.2 | 6.1 ± 0.5*** | 6.2 ± 0.4*** | 7.8 ± 0.4*** | −2 | 2 × 10−65 |
| Plateletcrit, % | 0.4 ± 0.1 | 0.4 ± 0.1 | 0.3 ± 0.1 | 0.3 ± 0.1 | 0.1 ± 0 | — | — |
| Red blood cells, 1012/L | 11.1 ± 0.7 | 10.5 ± 0.7 | 10.7 ± 0.8 | 10.4 ± 0.6 | 10.0 ± 0.6 | — | — |
| Hematocrit, % | 33.6 ± 2.4 | 32.7 ± 1.8 | 33.1 ± 2.5 | 34.1 ± 1.9 | 31.8 ± 1.7 | — | — |
| White blood cells, 109/L | 7.5 ± 3.6 | 8.6 ± 2.3 | 7.4 ± 2.6 | 10.1 ± 6.1 | 13.1 ± 6.8*** | 0.5 | 8 × 10−7 |
| Lymphocytes, 109/L | 6.2 ± 2.7 | 7.4 ± 2.0 | 6.9 ± 2.2 | 7.8 ± 3.7 | 11.8 ± 4.9*** | 0.5 | 3 × 10−11 |
| Monocytes, 109/L | 0.5 ± 0.4 | 0.3 ± 0.2 | 0.5 ± 0.2 | 0.3 ± 0.3 | 0.5 ± 0.4 | — | — |
| Neutrophils, 109/L | 0.8 ± 0.4 | 0.9 ± 0.4 | 0.7 ± 0.3 | 1.2 ± 0.8 | 1.7 ± 1.1 | — | — |
| Eosinophils, 109/L | 0.1 ± 0.2 | 0 ± 0 | 0 ± 0 | 0 ± 0.1 | 0 ± 0 | — | — |
| Basophils, 109/L | 0.1 ± 0.3 | 0 ± 0 | 0.1 ± 0.2 | 0 ± 0.1 | 0.1 ± 0.1 | — | — |
All values expressed as mean ± SD.
—, indicates that no statistical analysis was performed; λ, the coefficient for the power transformation to minimize heteroscedasticity and nonnormality; P, the result of a 1-way ANOVA on transformed data.
P < .001 compared with WT with the Dunnett post hoc test.
Reduced human G6b-B expression causes minor alterations in platelet function
G6bhu/hu platelets provide a unique model to investigate the physiological consequence of reduced human G6b-B levels in platelets, without altered expression of other receptors identified in G6b−/− mice.33 No significant defects were observed in αIIbβ3-mediated adhesion and spreading of G6bhu/hu platelets on fibrinogen, in the presence or absence of thrombin (supplemental Figure 8). Consistent with these findings, outside-in αIIbβ3 signaling was comparable between WT and G6bhu/hu platelets (supplemental Figure 9). Bands corresponding to tyrosine phosphorylated mouse (45 kDa) and human (28-34 kDa doublet) G6b-B were observed in fibrinogen-adhered WT and G6bhu/hu platelets, respectively. As G6bhu/hu mice do not express mouse G6b-B (Figure 3), the tyrosine phosphorylated band at 45 kDa (supplemental Figure 9) is due to the presence of other co-migrating phosphorylated proteins.
We next investigated (hemi)-ITAM receptor-mediated functional responses. Unexpectedly, platelet aggregation was marginally reduced in response to low and intermediate concentrations (1-10 μg/mL) of the GPVI-specific agonist collagen-related peptide (CRP) (Figure 4A). In contrast, CLEC-2-mediated platelet aggregation was marginally increased at 6 μg/mL anti-CLEC-2 antibody (Figure 4A). Platelet aggregation to thrombin, collagen, ADP and the TxA2 analog U46619 were normal (supplemental Figure 10). ATP secretion from dense granules was normal to all agonists tested (Figure 4A; supplemental Figure 10), except for a minor increase at an intermediate dose of collagen. These findings reveal minor alternations in platelet functional responses due to reduced human G6b-B expression that are likely of no physiological consequence.
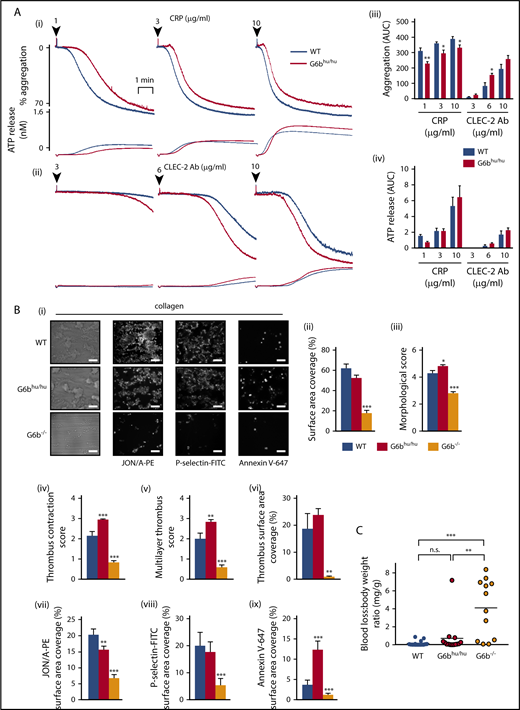
Minor alterations of G6bhu/huplatelet functional responses. (A) Averaged aggregation and ATP release traces for washed platelets (2 × 108/mL) activated with indicated concentrations of CRP (i) and anti-CLEC-2 antibody (Ab) (ii). Area under the curve (AUC) quantification of (iii) platelet aggregation and (iv) ATP release (n = 5-6 per condition, **P < .01 and *P < .05). (B) Heparin-PPACK-fragmin anticoagulated whole blood was flowed over glass coverslips coated with collagen immediately following collection. (i) Brightfield images were immediately collected, followed by staining with PE-conjugated JON/A, FITC-conjugated P-selectin, and Alexa 647–conjugated Annexin V and fluorescence imaging. Analysis quantifying (ii-vi) thrombus surface area coverage and morphological scores of adhered platelets, (n = 5-6; ***P < .001; **P < .01) and (vii-ix) surface area coverage of fluorescently labeled antibodies (n = 5-6; ***P < .001; *P < .05). (C) Total blood loss to body weight ratio of mice following excision of tail tip (n = 12-16; ***P < .001; **P < .01). All data collection and analysis in panels B and C were completed blinded, mean ± SEM. Scale bar, 10 μm.
Minor alterations of G6bhu/huplatelet functional responses. (A) Averaged aggregation and ATP release traces for washed platelets (2 × 108/mL) activated with indicated concentrations of CRP (i) and anti-CLEC-2 antibody (Ab) (ii). Area under the curve (AUC) quantification of (iii) platelet aggregation and (iv) ATP release (n = 5-6 per condition, **P < .01 and *P < .05). (B) Heparin-PPACK-fragmin anticoagulated whole blood was flowed over glass coverslips coated with collagen immediately following collection. (i) Brightfield images were immediately collected, followed by staining with PE-conjugated JON/A, FITC-conjugated P-selectin, and Alexa 647–conjugated Annexin V and fluorescence imaging. Analysis quantifying (ii-vi) thrombus surface area coverage and morphological scores of adhered platelets, (n = 5-6; ***P < .001; **P < .01) and (vii-ix) surface area coverage of fluorescently labeled antibodies (n = 5-6; ***P < .001; *P < .05). (C) Total blood loss to body weight ratio of mice following excision of tail tip (n = 12-16; ***P < .001; **P < .01). All data collection and analysis in panels B and C were completed blinded, mean ± SEM. Scale bar, 10 μm.
Altered G6bhu/hu platelet function does not affect thrombus formation or hemostasis
We further investigated G6bhu/hu platelet functional responses under physiological flow conditions on different thrombogenic surfaces. Platelet adhesion and thrombus formation were quantified by flowing anti-coagulated blood over: (1) collagen; (2) von Willebrand factor-binding protein (vWF-BP) and laminin; and (3) vWF-BP, laminin and the CLEC-2 agonist rhodocytin, for 3.5 minutes at 1000 s−1. Surface area coverage and morphological scores were used as measures of platelet adhesion and thrombus formation (supplemental Figure 11). Regression analysis of a cohort of WT mice (n = 19) did not indicate that the 21% reduction in platelet counts in G6bhu/hu mice will affect any of the measured parameters (supplemental Figure 12). The total surface area covered by G6bhu/hu platelets on collagen was unaltered compared with WT platelets (Figure 4B). However, multilayer thrombus formation and contraction were both significantly increased, despite a minor reduction in αIIbβ3 activation and normal α-granule release, measured by immunostaining with JON/A and P-selectin antibodies, respectively (Figure 4B). Altered thrombus morphology of G6bhu/hu platelets is most likely a reflection of the increased proportion of phosphatidylserine (PS)-positive procoagulant platelets, measured by Annexin V staining (Figure 4B).
No difference in adhesion or platelet activation were observed when anti-coagulated blood from WT and G6bhu/hu mice was flowed over vWF-BP and laminin, or vWF-BP, laminin and rhodocytin (supplemental Figure 13). Although some minor alterations in G6bhu/hu platelet activation were identified using this assay, expression of human G6b-B rescued the severe defects observed in G6b−/− platelets. This rescue is likely due to a combination of increased platelet counts and normalized platelet reactivity. Functional defects observed in G6bhu/hu platelets did not culminate in compromised hemostasis in mice, as assessed using the tail bleeding assay, whereas G6b−/− mice bled excessively, as previously reported (Figure 4C).33
To determine the molecular basis of altered responses to CRP and anti-CLEC-2 antibody, we investigated tyrosine phosphorylation in WT and G6bhu/hu platelets. The findings from aggregations studies were supported by a minor reduction in global tyrosine phosphorylation following CRP stimulation (Figure 5A). A trend toward reduced SFK and Syk activities was identified, as measured by changes in autophosphorylation of Tyr418 and Tyr519/20 in the activation loops of SFKs and Syk, respectively, though this did not reach significance (Figure 5A). SFK-mediated phosphorylation of the ITAM-containing FcR γ-chain, which acts as a docking site for Syk to propagate signals downstream of GPVI, was also marginally reduced in CRP-stimulated G6bhu/hu platelets. The opposite was observed in CLEC-2 activated platelets, in which global tyrosine phosphorylation, SFK and Syk activation were marginally increased in response to antibody-mediated cross-linking and activation of CLEC-2 (Figure 5B). However, only Syk activation with 10 μg/mL anti-CLEC-2 antibody was significantly increased.
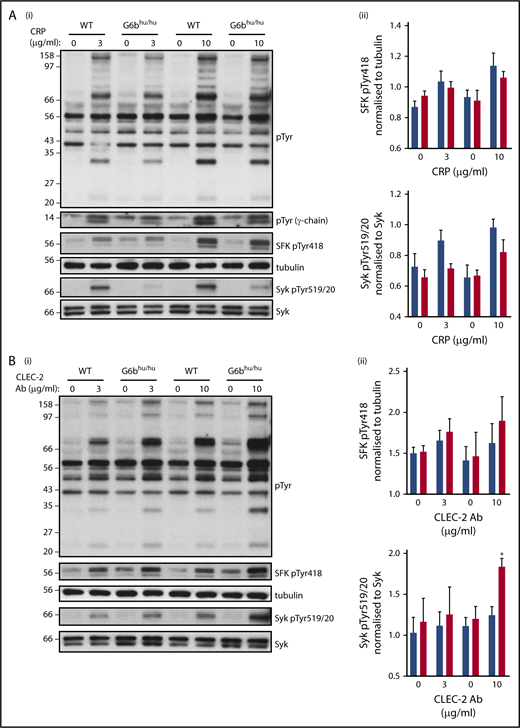
Altered tyrosine phosphorylation in response to GPVI and CLEC-2 agonists in G6bhu/huplatelets. Representative blots (n = 3) of lysates prepared from washed platelets (4 × 108/mL) activated with indicated concentrations of (Ai) CRP (90 seconds, 10 μM lotrafiban, 10 μM indomethacin, and 2 U/mL apyrase) or (Bi) CLEC-2 Ab (300 seconds, 10 μM lotrafiban) and probed with the indicated antibodies. (Aii,Bii) Src family kinase (SFK) phosphotyrosine 418 (pTyr418) and (Aiii,Biii) Syk pTyr519/20 were quantified using ImageJ and normalized to total tubulin and Syk reblots, respectively. WT, blue; G6bhu/hu, red.
Altered tyrosine phosphorylation in response to GPVI and CLEC-2 agonists in G6bhu/huplatelets. Representative blots (n = 3) of lysates prepared from washed platelets (4 × 108/mL) activated with indicated concentrations of (Ai) CRP (90 seconds, 10 μM lotrafiban, 10 μM indomethacin, and 2 U/mL apyrase) or (Bi) CLEC-2 Ab (300 seconds, 10 μM lotrafiban) and probed with the indicated antibodies. (Aii,Bii) Src family kinase (SFK) phosphotyrosine 418 (pTyr418) and (Aiii,Biii) Syk pTyr519/20 were quantified using ImageJ and normalized to total tubulin and Syk reblots, respectively. WT, blue; G6bhu/hu, red.
Further reduction in human G6b-B expression exacerbates platelet defects
To further investigate the threshold of human G6b-B expression required to rescue the G6b−/− phenotype, we generated human G6b hemizygous mice (G6bhu/−) by crossing G6bhu/hu mice with G6b−/− mice (Figure 6A). G6bhu/− mice expressed only 12% of human G6b-B and -A compared with human platelets, and no mouse G6b-B (Figure 6A; supplemental Figure 14A, B). These expression levels produced an even greater reduction in platelet counts (29%) and a comparable increase in platelet volume (6% to 7%) compared with G6bhu/hu mice (Table 2). This provides further evidence that human G6b-B dose dependently regulates platelet production.
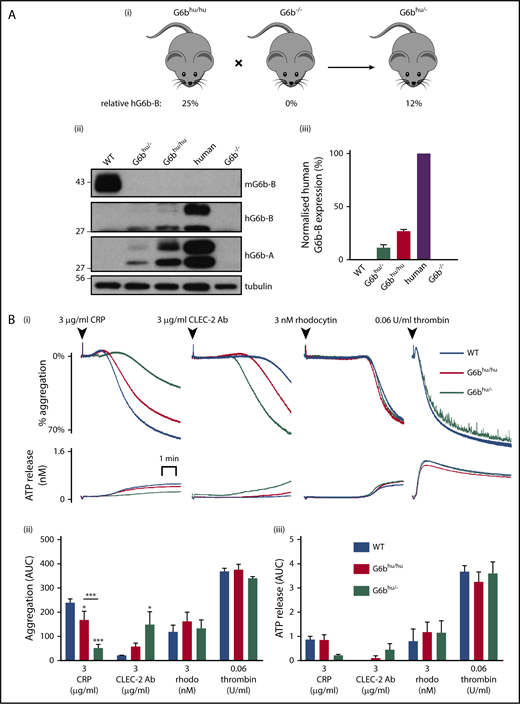
Further reduction in human G6b-B expression exacerbates observed phenotype. (Ai) G6bhu/hu mice were crossed with G6b−/− mice to produce hemizygous (G6bhu/−) mice. (Aii) Representative blots of mouse G6b-B (mG6b-B), human G6b-B (hG6b-B) and hG6b-A and (Aiii) quantification of hG6b-B expression in the indicated genotypes (n = 3). (B) Average aggregation and ATP release traces (Bi) and quantification of AUC (Bii,iii) investigating functional response of G6bhu/− mouse washed platelets (2 × 108/mL) in response to indicated agonists (n = 5-6; ***P < .001; *P < .05). All bar graphs presented as mean ± SEM.
Further reduction in human G6b-B expression exacerbates observed phenotype. (Ai) G6bhu/hu mice were crossed with G6b−/− mice to produce hemizygous (G6bhu/−) mice. (Aii) Representative blots of mouse G6b-B (mG6b-B), human G6b-B (hG6b-B) and hG6b-A and (Aiii) quantification of hG6b-B expression in the indicated genotypes (n = 3). (B) Average aggregation and ATP release traces (Bi) and quantification of AUC (Bii,iii) investigating functional response of G6bhu/− mouse washed platelets (2 × 108/mL) in response to indicated agonists (n = 5-6; ***P < .001; *P < .05). All bar graphs presented as mean ± SEM.
Surface receptor expression was normal in G6bhu/− platelets compared with WT platelets, except for CLEC-2, which was marginally reduced by 16% (supplemental Figure 14C). Interestingly, platelet aggregation was further reduced in response to CRP and increased in response to antibody-mediated cross-linking of CLEC-2 (Figure 6B). These findings demonstrate differential roles of G6b-B in regulating ITAM- and hemi-ITAM-containing receptor-mediated platelet activation at these expression levels. No difference was observed upon activation of CLEC-2 using the snake toxin rhodocytin. This likely reflects the tetravalent structure of rhodocytin that can elicit a more robust CLEC-2 activating signal, thus abrogating the mild phenotype observed with the bivalent anti-CLEC-2 antibody. Thrombin-mediated aggregation of G6bhu/− platelets was normal and no significant defects in ATP secretion was observed for all agonists tested.
Discussion
Here we have described four families with a distinct phenotype of macrothrombocytopenia, microcytic anemia, variable leukocytosis and histologically characteristic megakaryocytic clustering, dysplasia and focal reticulin fibrosis in the BM presenting at young age. We attribute this to homozygous loss-of-function mutations in G6b, the gene encoding G6b-B, a megakaryocytic/platelet-specific ITIM-containing receptor. While this work was in preparation, another consanguineous Arab family with a similar phenotype was described with a homozygous predicted null mutation in G6b.26 These data firmly support the conclusion that recessive loss-of-function mutations in G6b result in the phenotype that we comprehensively describe and term congenital macrothrombocytopenia with cMTFM. All of the clinically affected patients have a phenotype strikingly similar to a null mutation,33 or mutations that abolish ITIM/ITSM signaling (Geer et al)34 in the murine ortholog. Concomitantly, we have demonstrated that the G6b KO phenotype can be rescued upon expression of human G6b.
One apparently clinically unaffected individual, 1-III-9, without thrombocytopenia or anemia at the time of her evaluation as a transplant donor, also shares the G6b-B mutation; she did not have thrombocytopenia, bleeding symptoms, leukocytosis, anemia, or atypical megakaryocytes associated with fibrosis, but did have rare large platelets. She did have transient BM lymphoid aggregates that are unusual for such a young child; these might be indicators of a primary BM disorder, but could equally be due to her CAH or other causes. Thus, it is likely that even genotypically similar patients might have varying expressivity or penetrance of the phenotype due to other modifying factors, including age, environment, and genotypes at other loci. Indeed, for example, patient 2-II-5 came to medical attention only because of his more severely affected younger sibling, 2-II-6. In addition to modifiers noted above, there are several possible explanations for the successful HSCT of the two siblings, 1-III-5 and 1-III-6 from their cousin, 1-III-9, who shares the same homozygous mutant G6b genotype. First, it is conceivable that the conditioning regimen alters the niche directly, which could impact the regulation of other components important in the regulation of megakaryocytic development, proplatelet and platelet formation. Second, introduction of a new hematopoietic stem cell pool from the donor into the host with cMF could lead to immunological graft and host interactions between the stem cell compartment and the niche that could alter megakaryocytic development.
The cMTFM phenotype underscores the importance of G6b-B signaling in normal megakaryocyte and platelet biology. Binding of Shp1 and Shp2 are essential for mediating signal transduced by G6b-B. A mutant form of G6b-B that uncouples it from Shp1 and Shp2 signaling phenocopies the G6b KO mouse model (Geer et al),33,34 and a tissue-specific mouse model of combined Shp1 and Shp2 deficiency recapitulates multiple features of G6b KO animals,44 further indicating the essential nature of this signaling pathway to megakaryocyte function.
The majority of reported patients with cMF have undergone HSCT as it was felt to be the only curative treatment optio.15 The histological resemblance to PMF in adults, which carries a risk of evolution to acute myeloid leukemia and a poor prognosis, may have encouraged providers to offer HSCT out of due caution, even in the absence of evidence for adverse outcomes when managed conservatively. Indeed, in our series of 7 cases of patients with G6b mutations, three required HSCT for cytopenias, one was clinically unaffected, and three have been observed without ongoing clinical interventions. The identification of a molecular therapeutic target—Shp1- and Shp2-dependent signal transduction—now offers the potential to circumvent the G6b-B deficiency in cMTFM and insights into the pathogenesis of other forms of primary and secondary myelofibrosis.
The humanized G6b mouse provided compelling evidence of the analogous functions of human and mouse G6b-B. The mild platelet phenotype, suggests human G6b-B interacts with the same ligands and signals in a similar manner to mouse G6b-B. Reduced binding of human G6b-B to mouse Shp1 is most likely a reflection of the relative stoichiometries and binding affinities of Shp1 and Shp2 in mouse platelets. Mouse platelets contain 6-fold higher levels of Shp2 than Shp1, whereas human platelets contain 2-fold more Shp1 than Shp2 (supplemental Figure 3).42,43 In addition, the binding affinity of tandem SH2 domains of human Shp2 with phosphorylated-G6b-B-ITIM-ITSM peptide is 100-fold greater than the binding affinity of tandem SH2 domains of Shp1.45 However, differences in the proportions of human G6b-B bound Shp1 and Shp2 in mouse versus human platelets are not likely to underlie the platelet defects described the humanized G6b mouse, as mouse G6b-B is predicted to bind the same ratios of Shp1 and Shp2. Thus, platelet defects are more likely due to reduced human G6b-B expression.
Despite the significant reduction in human G6b-B in G6bhu/hu and G6bhu/− mice, both mouse models exhibited only mild platelet phenotypes. A large proportion of G6b-B is therefore not essential for maintenance of platelet homeostasis. MKs from G6b−/− mice were previously shown to develop normally in vitro, but with significantly diminished proplatelet production capacity.33 Reduced platelet production in G6bhu/hu and G6bhu/− mice could therefore have been as a consequence of aberrant proplatelet formation, arising from reduced G6b-B expression in MKs. The dose dependent nature of the reduction in platelet counts in these homozygous and heterozygous mice adds to the growing body of evidence of the vital role of G6b-B in regulating platelet production.
The reduction in human G6b-B expression was predicted to lead to an increase in platelet reactivity to collagen. Indeed, thrombus formation and contraction were significantly increased when whole blood was flowed over collagen, despite a marginal reduction in integrin αIIbβ3 activation. This can be explained by an enhancement in PS exposure and increased procoagulant activity of G6bhu/hu platelets, which is dependent upon PLCγ2-mediated Ca2+ flux and phosphatidylinositol 3-kinase signaling,45 highlighting these as potential downstream targets of Shp1 and Shp2. The dose-dependent reduction in aggregation of G6bhu/hu and G6bhu/− platelets to CRP was however, counter-intuitive, suggesting G6b-B may act as both a positive and negative regulator of GPVI signaling, depending upon expression levels. The candidate target of G6b-B-associated Shp1 and Shp2 is Syk tyrosine kinase that we previously showed is hyper-activated in G6b-B-deficient platelets.33 Syk contains multiple regulatory tyrosine phosphorylation sites that Shp1 and Shp2 may differentially dephosphorylate, depending upon the expression level of G6b-B, leading to either positive or negative effects on Syk activity.47
This study establishes that human and mouse G6b-B perform the same physiological functions, and that deletion and loss-of-function mutations in G6b-B lead to megakaryocytic and myelofibrotic disorders in both species. Validation of these analogous functions is essential to progress with exploring the role of G6b-B in various pathological conditions in humans and as a novel therapeutic drug target. The humanized G6b-B mouse model described here provides an invaluable tool for investigating the efficacy and therapeutic potential of agents targeting human G6b-B in various disease conditions, including myeloproliferation and myelofibrosis, allowing for a greater understanding of the mechanism of action of these therapies.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
Sample collection and clinical and pathology analysis were performed under the Pediatric MDS and BMF Registry supported by National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases grant R24 DK 099808. I.H. and M.D.F. were supported by National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases grant R24 DK099808. M.J.G. was funded by a Medical Research Council studentship (GBT1564). T.V. was funded by the Deutsche Forschungsgemeinschaft (DFG V2134-1/1).
A.M. is a British Heart Foundation (BHF) Intermediate Basic Science Research Fellow (FS/15/58/31784) and Y.A.S. is a BHF Senior Basic Science Research Fellow (FS/13/1/29894).
Authorship
Contribution: I.H. identified patients, analyzed the clinical and pathologic phenotypes, designed the study, and wrote and revised the manuscript; M.J.G. performed experiments, analyzed data, and wrote and revised the manuscript; T.V. and J.P.v.G. performed experiments, analyzed data, and revised the manuscript; A.C. and D.R.C. performed patient sample processing and sequencing analysis; A.B., S.H., M.J.E.K., J.W.M.H., and M.D. performed experiments and analyzed data; M.L.C. developed the customized immunohistochemistry for G6b-B; J.A.E. provided snake toxin rhodocytin; K.S.-A. and K.M. performed linkage and WES data analysis; E.A.O. provided patient care; K.F. performed GWAS; C.P. and R.F. identified and diagnosed patients; G.E.J. and E.T. analyzed data and revised the manuscript; W.H.O. analyzed data and contributed intellectually; A.M. designed experiments, analyzed data, and revised the manuscript; M.D.F. analyzed the clinical and pathologic phenotypes, designed the study, and wrote and revised the manuscript; Y.A.S. conceptualized, designed experiments, analyzed data, and wrote and revised the manuscript; and all authors contributed to the revision of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Inga Hofmann, University of Wisconsin, Madison, UW Health, WIMR 4151, 1111 Highland Ave, Madison, WI 53705; e-mail: ihofmann@wisc.edu; and Yotis A. Senis, University of Birmingham, Birmingham, United Kingdom, B15 2TT; e-mail: y.senis@bham.ac.uk.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal