

In this issue of Blood, noted that mutations in signal recognition particle 54 (SRP54) cause an inherited neutropenia in 23 individuals with features of both severe congenital neutropenia (SCN) and Shwachman-Diamond syndrome (SDS).1
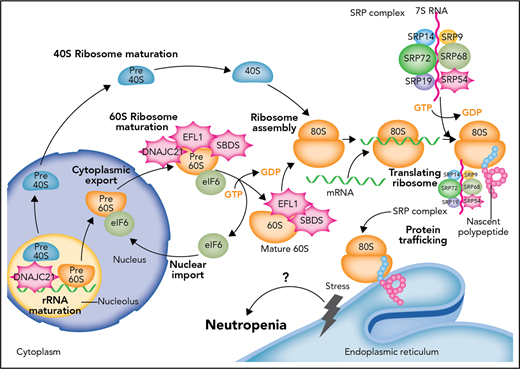
Defects in the proteostasis pathway lead to inherited neutropenia. Molecular cloning of genetic lesions in individuals with moderate to severe neutropenias have centered on the biosynthetic pathway for proteins and their folding and trafficking (proteostasis for protein homeostasis). The most common genetic lesion in SDS is the biallelic mutation of SBDS. SBDS interacts with EFL1 to displace eukaryotic initiation factor 6 (eIF6). DNAJC21 stabilizes the 60S ribosome. As part of the SRP complex, SRP54 escorts the nascent polypeptide to the ER to complete translation and possible posttranslational modification. When defects arise in this prodigious and continuous process, unfolded protein response and ER stress follows. The most commonly mutated gene in SCN is ELANE, which encodes a serine protease. It is thought that mutated ELANE results in proteins that misfold and cause unfolded protein response. Through unknown mechanisms of apoptosis and/or differentiation impairment, granulopoiesis cannot be completed in SCN or sufficiently produced in SDS. How SCN and SDS transform frequently to MD or AML remains inadequately understood. GDP, guanosine diphosphate; GTP, guanosine triphosphate. Professional illustration by Somersault18:24.
Defects in the proteostasis pathway lead to inherited neutropenia. Molecular cloning of genetic lesions in individuals with moderate to severe neutropenias have centered on the biosynthetic pathway for proteins and their folding and trafficking (proteostasis for protein homeostasis). The most common genetic lesion in SDS is the biallelic mutation of SBDS. SBDS interacts with EFL1 to displace eukaryotic initiation factor 6 (eIF6). DNAJC21 stabilizes the 60S ribosome. As part of the SRP complex, SRP54 escorts the nascent polypeptide to the ER to complete translation and possible posttranslational modification. When defects arise in this prodigious and continuous process, unfolded protein response and ER stress follows. The most commonly mutated gene in SCN is ELANE, which encodes a serine protease. It is thought that mutated ELANE results in proteins that misfold and cause unfolded protein response. Through unknown mechanisms of apoptosis and/or differentiation impairment, granulopoiesis cannot be completed in SCN or sufficiently produced in SDS. How SCN and SDS transform frequently to MD or AML remains inadequately understood. GDP, guanosine diphosphate; GTP, guanosine triphosphate. Professional illustration by Somersault18:24.
In fact, after ELANE, mutations in SRP54 are the second most common cause of inherited neutropenia in the French Congenital Neutropenia Registry. Together with the 3 patients reported by Carapito et al,2 the work of Bellanné-Chantelot et al adds to our understanding of genetic pathways and phenotype correlations of inherited neutropenias, but it raises the question of how to classify these and other genetic cytopenias: molecular pathway-based classification or phenotype-based classification?
The inherited bone marrow failure syndromes constitute a heterogeneous group of blood disorders that are phenotypically distinct.3 They are true experiments of nature, enlightening our understanding of normal and pathologic hematopoiesis and often non-hematopoietic tissue development. Their importance goes further with their acting as leukemia and/or cancer predisposition syndromes. Molecular cloning has reinforced phenotype distinctions by revealing that mutations in pathways lead to the separate clinical entities. Fanconi anemia is caused by at least 20 different genes involved in DNA damage response, Diamond-Blackfan anemia by at least 15 genes involved in ribosomal structure, and dyskeratosis congenita by at least 10 genes that promote telomere maintenance and stability.
The inherited neutropenias include SCN, cyclic neutropenia (CyN), SDS, and a motley group of monogenic disorders.4 SCN results from more than 8 different genes, which encode a variety of proteins that have defied easy categorization. Approximately 50% of patients with SCN harbor mutations in the neutrophil serine protease ELANE. Individuals with SCN typically have recurrent life-threatening bacterial infections beginning during infancy. The absolute neutrophil count (ANC) is chronically <200 cells per μL. Recombinant human granulocyte colony-stimulating factor (G-CSF [filgrastim]) improves the severe neutropenia and dramatically reduces the occurrence of infections. For SCN patients, there is concern that filgrastim may accelerate the transformation of SCN to myelodysplastic syndrome (MDS) or acute myeloid leukemia (AML) because of acquired mutations in CSF3R (G-CSF receptor). CyN is another inherited disorder characterized by severe neutropenia that occurs every 21 days (the nadir may occur in 14- to 35-day cycles). Occasionally, erythrocyte and platelet numbers display cyclic variations. Almost all of the CyN is a result of germ line ELANE mutations. These individuals typically present in the first decade of life or later, with less severe or frequent infections but annoying mouth sores that resolve when the neutropenia abates.
SDS is characterized by short stature and thin habitus, neutropenia, pancreatic insufficiency, and skeletal anomalies. The neutropenia is classically moderate (ANC >500 cells per μL) and can fluctuate. A minority of patients have infections and/or depend on filgrastim to maintain ANC >750 cells per μL. SDS patients also have a high risk for transformation to MDS or AML. In up to 90% of patients, SDS results from biallelic mutations in the eponymous SBDS gene that encodes an assembly factor for the 80S ribosome. Mutational gene discoveries over the past 2 years in individuals who seem to have SDS but actually have wild-type SBDS have been identified: the chaperone DNAJC21,5 the elongation factor-like GTPase 1 (EFL1),6 and now SRP54. All these proteins are, in one way or another, involved in protein synthesis: ribosomal RNA (rRNA) or ribosomal subunit maturation and protein trafficking to the endoplasmic reticulum (ER). SBDS interacts with EFL1 on the pre-60S ribosome to promote assembly of the 80S ribosome from combining the 40S and 60S subunits, DNAJC21 is implicated in rRNA biogenesis and late cytoplasmic maturation of the 60S subunit, and SRP54 helps to direct proteins coming off the 80S ribosome and transfer them to the ER for correct trafficking. The signal recognition particle (SRP) itself is a ribonucleoprotein complex of 6 proteins: SRP9, SRP14, SRP19, SRP54, SRP68, and SRP72 (see figure). SRP72 has been associated with familial aplastic and myelodysplastic syndromes.7
Bellanné-Chantelot and colleagues showed that SRP54 mutants lead to ER stress and autophagy. Activation of the unfolded protein response pathways has also been described in patients who have SCN with ELANE mutations. They also observed a high reduction of proliferation of granulocytic cells and apoptosis dependent on the P53 pathway, characteristics also observed in patients with SDS and Diamond Blackfan anemia. Unlike SCN or SDS, none of the patients with SRP54 mutations (23 cases) transformed to leukemia, despite high doses of filgrastim, after a median follow-up of 15 years. Unlike in SCN or SDS, none of the 17 patients analyzed by deep sequencing harbored mutations in CSF3R, RUNX1, or TP53, which may be associated with risk of leukemogenesis.8 How defects in these different pathways and cellular stress responses cause neutropenia that frequently transforms to AML or MDS is still not well understood. Might the risk of leukemic transformation and the identities of cooperating mutants exist in mutant SRP54-associated neutropenia? Must they await their emergence with a longer follow-up?
The title of Bellanné-Chantelot’s article suggests ambivalence regarding which class of inherited neutropenia SRP54 mutations fit in: SCN, SDS, or both. In this series, the median ANC was 220 cells per μL, and the use of filgrastim was high (87%) but not universal, unlike with SCN. In this series, only 2 individuals had exocrine pancreatic insufficiency, unlike with SDS. Six (of 23) had clinically significant neurologic abnormalities. Morphologically, there was a promyelocytic arrest, like with SCN and unlike with SDS. Dysplastic features were common, perhaps more like SDS.9
Classification of human diseases has advanced considerably since the ancient Greeks divided them mechanistically into the four humors. Next-generation sequencing has offered both precision and complexity to hematologic diseases. Should one lump or split syndromes? Even in monogenically defined blood diseases (eg, sickle cell anemia, hemophilia A, or SBDS-mutated SDS), how does one account for phenotypic diversity? Could a subatomic genetic classification be looming? Apropos, does an SRP54 mutation associated with neutropenia belong to SCN or SDS? Can there be a useful classification scheme for the inherited neutropenias, the list of which grows seasonally?
Conflict-of-interest disclosure: The authors declare no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal