

Key Points
Identification of SRP54 mutations in congenital neutropenia.
SRP54 mutations induce ER stress and autophagy associated with apoptosis.

Abstract
Congenital neutropenias (CNs) are rare heterogeneous genetic disorders, with about 25% of patients without known genetic defects. Using whole-exome sequencing, we identified a heterozygous mutation in the SRP54 gene, encoding the signal recognition particle (SRP) 54 GTPase protein, in 3 sporadic cases and 1 autosomal dominant family. We subsequently sequenced the SRP54 gene in 66 probands from the French CN registry. In total, we identified 23 mutated cases (16 sporadic, 7 familial) with 7 distinct germ line SRP54 mutations including a recurrent in-frame deletion (Thr117del) in 14 cases. In nearly all patients, neutropenia was chronic and profound with promyelocytic maturation arrest, occurring within the first months of life, and required long-term granulocyte colony-stimulating factor therapy with a poor response. Neutropenia was sometimes associated with a severe neurodevelopmental delay (n = 5) and/or an exocrine pancreatic insufficiency requiring enzyme supplementation (n = 3). The SRP54 protein is a key component of the ribonucleoprotein complex that mediates the co-translational targeting of secretory and membrane proteins to the endoplasmic reticulum (ER). We showed that SRP54 was specifically upregulated during the in vitro granulocytic differentiation, and that SRP54 mutations or knockdown led to a drastically reduced proliferation of granulocytic cells associated with an enhanced P53-dependent apoptosis. Bone marrow examination of SRP54-mutated patients revealed a major dysgranulopoiesis and features of cellular ER stress and autophagy that were confirmed using SRP54-mutated primary cells and SRP54 knockdown cells. In conclusion, we characterized a pathological pathway, which represents the second most common cause of CN with maturation arrest in the French CN registry.
Introduction
Congenital neutropenia (CN) is a heterogeneous group of rare genetic disorders that have in common an impaired maturation of neutrophil granulocytes.1 Some forms of CN are restricted to the hematological defect, whereas others are syndromic and associated with extrahematopoietic organ dysfunctions affecting the pancreas, brain, heart, bone, and skin. Some patients with CN present a high risk for leukemic transformation.
Molecular abnormalities in 24 genes have been identified in CN.1 In about 25% of patients with a clinical history suggestive of CN, the genetic defects remain unknown.2 The pathways linked to the genetic defects of CN involve cellular stress mechanisms,3,4 including unfold protein response (ELANE),5,6 endoplasmic reticulum (ER) stress (G6PC3, JAGN1),7,8 defective endosome trafficking (VPS13B, VPS45),9,10 impaired intracellular glucose homeostasis (G6PC3),7 and defective ribosome biogenesis (SBDS, DNAJC21, EFL1).11-13 Of note, these different pathways result in an increased apoptosis of neutrophil granulocytes and of their precursors.3 Further elucidations of the molecular mechanisms that cause CN are important to extend our understanding of this heterogeneous disease.
Here, we report that the 54-kDa signal recognition particle (SRP54) GTPase protein, a key component of the ribonucleoprotein complex that mediates the cotranslational targeting and the translocation of secretory and membrane proteins to the ER,14,15 is involved in the granulocytic differentiation of myeloid precursor cells. We showed that an impaired SRP54 pathway leads to enhanced ER stress, autophagy, and apoptosis of neutrophil granulocytes and of their precursors. We describe the molecular, clinical, and functional investigations of 23 cases of CN linked to the SRP pathway.
Methods
Subjects and samples
Informed consents from the individuals or their parents, for children included in the study, were obtained according to the Declaration of Helsinki. All material (blood, bone marrow, primary fibroblasts, and DNA samples) were declared to French Health Authorities in compliance with current legislation. All the individuals with CN were registered with informed assent in the French severe chronic neutropenia registry (CNIL certificate 97.075). Hematologic parameters at diagnosis and during follow-up, bone marrow cell counts morphology (centrally reviewed by OF), bacterial infections, dose and frequency of granulocyte colony-stimulating factor (G-CSF) treatment, extrahematopoietic symptoms, and family history were recorded in the registry.
Material
Liquid culture media, including Iscove’s Modified Dulbecco Medium, were from Invitrogen. Human recombinant stem cell factor (SCF) was a generous gift from Biovitrum AB. Interleukin 3 (IL-3) was from Miltenyi Biotec. Nutlin 3a was a gift from L. T. Vassilev (Roche). The acute myeloblastic leukemia HL-60 cell line and derivatives (expressing shRNA) were cultured in RPMI supplemented with 10% fetal bovine serum.
Whole-exome sequencing
Whole-exome sequencing (WES) was performed from genomic DNA extracted from peripheral blood sample, using the SureSelect XT Clinical Research Exome-54 Mb (Agilent). Library preparation, exome sequencing, and row data analysis were performed by IntegraGen SA. Sequence reads were mapped to the human genome build hg19/GRCh37. Detailed information on WES procedure and data analysis are provided in supplemental Data, available on the Blood Web site.
Sanger sequencing
SRP54 gene (NM_003136.3) sequencing was performed using standard procedures. Primers are described in supplemental Table 1. Sequences were analyzed with Seqscape software v2.5 (Life Technologies), and variants were annotated with Alamut software v2.7 (Interactive Biosoftware).
ShRNA lentiviral constructs and production of viral particles
Lentiviral short hairpin (sh)RNA strategy was used to knockdown SRP54 expression, as previously described.16 Sequences for human shRNA-SRP54 are described in supplemental Table 1. Lentiviral particles containing pRRLsin-PGK-eGFP-WPRE vector (Genethon) were produced as previously described.16 HL-60 or isolated human progenitors were transduced with lentivirus and sorted on GFP+ 72 hours later on a Becton Dickinson (BD) Influx cell sorter.
CD34+ cell isolation and cultures
CD34+ cells were obtained from leukapheresis samples after mobilization of donors or from peripheral blood or bone marrow from patients with CN. CD34+ cells were isolated by a positive selection using an immunomagnetic cell sorting system (Miltenyi Biotec). They were transduced or not with shRNA, GFP+-sorted, and cultured in serum-free medium17 supplemented with SCF (25 ng/mL), IL-3 (10 ng/mL), and G-CSF (20 ng/mL) for 21 days. Selected populations were sorted for granulocytic markers between days 10 and 14.
Flow cytometry analysis
Conjugated anti-CD15-V450, anti-CD11b-PE, and anti-CD36-APC monoclonal antibodies (BD) were used and the cells were either analyzed for surface markers or sorted on a BD Influx cell sorter. For apoptosis analyses, cells were stained with Annexin V-APC (BD) and propidium iodide (Sigma).
Transmission electron microscopy
Cells were fixed in 2% glutaraldehyde in 0.1 M phosphate buffer for 1 hour at 4°C, and postfixed in 2% osmium tetroxide. After ethanol dehydration, cell pellets were embedded in Epon812. Ultrathin sections were stained with standard uranyl acetate and lead citrate before observation with a Tecnai 12 electron microscope (FEI). Digital images were taken with a SISMegaviewIII charge-coupled device camera (Olympus).
Western blot
Signaling studies were performed on day 10-granulocytic precursors by in vitro culture of CD34+ progenitors. Phosphorylation status of STAT3 (Tyr 705), ERK1/2 (Thr 202/Tyr 204), AKT (Ser 473), PERK, eiF2a, ULK1, the pan proteins (Cell Signaling Technology), and β-Actin (Sigma) were checked by western blot analysis.
Real-time quantitative reverse transcription polymerase chain reaction
Polymerase chain reactions were carried out in the ABI Prism GeneAmp 5700 (Perkin-Elmer), using the Power SYBR-Green PCR Master Mix (ABI) containing the specific primers (supplemental Table 1). The expression levels of all genes were calculated relatively to HPRT and to PPIA housekeeping genes using qbase+ software.
Results
Identification of germ line mutations in SRP54 in CN
We focused our study on patients with a profound and permanent neutropenia defined by an absolute neutrophil count less than 0.5 × 109 G/L associated with a promyelocytic maturation arrest and diagnosed in the neonate period or during childhood. Immune-mediated causes as well as the most frequent genetic etiologies associated with CN were excluded (data not shown). We selected 8 sporadic cases and 6 multiplex families, including 2 with an autosomal dominant inheritance and 4 compatible with a recessive inheritance. WES analysis was performed on a trio-based approach. Focusing first on the search for de novo heterozygous coding variants in the sporadic cases, we identified a unique mutation in the patient P19/F15, whereas 3 and 24 variants were, respectively, found in the P11/F10 and P13/F12 patients (Table 1; supplemental Table 2). Interestingly, heterozygous mutations of the same gene, SRP54, were present in the 3 probands. These were a missense mutation (c.407G>A, p.Cys136Tyr) in patient P19/F15 and the same in-frame deletion (c.349_351del, p.Thr117del) in P11/F10 and P13/F12 patients (Figure 1A). SRP54 was an attractive candidate because of its high expression in myeloid cells and its key role in targeting membrane and secretory proteins to the ER.18,19 The subsequent WES analysis of the multiplex families revealed a distinct missense mutation (c.353G>A, p.Cys118Thr) of SRP54 in the 2 affected patients (P16, P17) of the family F14. SRP54 mutations were confirmed by Sanger sequencing of DNA extracted from blood and fibroblasts (Figure 1A).
Clinical and molecular characteristics of individuals with SRP54 mutations
| Family | ID | Sex | Age at diagnosis | Age at last follow-up (years) | ANCs (per microliter) before G-CSF therapy | Severe bacterial infections* | Stomatitis gingivitis | Maturation arrest on bone marrow smear (before G-CSF therapy) | G-CSF therapy mean (maximum) dose (µg/kg/day) | Neurological symptoms | Exocrine pancreatic insufficiency† | Other extrahematopoietic symptoms | SRP54 genotype cDNA; protein inheritance |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| F1 | P1 | M | 9.8 months | 8 | 270 | Yes | Yes | Yes | Yes/5 (5) | No | No | No | c.337G>C p.Gly113Arg de novo |
| F2 | P2 | M | 4.6 months | 3 | 443 | Yes | No | Yes | Yes/16 (20) | No | Yes | No | c.349_351del p.Thr117del de novo |
| F3 | P3 | F | 1.7 months | 15 | 440 | Yes | Yes | Yes | Yes/4 (5) | No | No | No | c.349_351del p.Thr117del dominant |
| P4 Father | M | 2.1 years | 44 | 100 | No | Yes | Yes | No | No | No | No | ||
| F4 | P5 | M | 10.6 months | 11 (HSCT at 1.5) | 190 | Yes | No | Yes | Yes/refractory till 50 µg/kg | Intellectual disability, autism spectrum disorder‡ | No | No | c.349_351del p.Thr117del nd |
| F5 | P6 | M | 5 years | 32 | 530 | No | Yes | Yes | Yes/16 (20) | No | No | Osteoporosis Type 2 diabetes at 31 years | c.349_351del p.Thr117del de novo§ |
| F6 | P7 | M | 2 days | 10 | 45 | Yes | Yes | Yes | Yes/6 (15) | No | No | No | c.349_351del p.Thr117del de novo |
| F7 | P8 | F | 10.1 months | 15 | 120 | No | Yes | Yes | Yes/10 (10) | No | No | No | c.349_351del p.Thr117del de novo |
| F8 | P9 | M | 1 months | 24 | 250 | Yes | Yes | Yes | Yes/5 (5) | No | No | IUGR, GH deficiency and GH therapy (Final height of 1.69 m) | c.349_351del p.Thr117del nd |
| F9 | P10 | M | 4.2 months | 11 | 230 | Yes | No | Yes | Yes/10 (10) | No | No | No | c.349_351del p.Thr117del de novo |
| F10 | P11‖ | M | 12.7 months | 7 | 360 | Yes | Yes | Yes | Yes/5 (5) | No | No | No | c.349_351del p.Thr117del de novo |
| F11 | P12‖ | M | 1.3 years | 39 | 90 | Yes | Yes | Yes | Yes/2 (5) | No | No | No | c.349_351del p.Thr117del de novo |
| F12 | P13‖ | F | 1 days | 3 | 100 | Yes | N | Yes | Yes/20 (20) | No | No | No | c.349_351del p.Thr117del de novo |
| F13 | P14 | M | 10 days | 6 | 110 | Yes | Yes | Yes | Yes/5 (5) | No | No | No | c.349_351del p.Thr117del dominant |
| P15 | F | 20 years | 28 | 345 | No | Yes | Yes | No | No | No | No | ||
| F14 | P16‖ | F | 4.2 months | 17 | 435 | Yes | No | Yes (intermittent) | Yes/5 (5) | No | No | No | c.353G>A p.Cys118Tyr dominant |
| P17‖ Father | M | 2.8 months | 46 | 960 | Yes | Yes | Yes (intermittent) | Yes/5 (5) | Learning difficulties** | No | No | ||
| P18 Brother | M | 8.3 months | 21 | 1090 | No | No | No | No | Learning difficulties** | No | No | ||
| F15 | P19‖ | F | 24 days | 6 | 215 | Yes | Yes | Yes | Yes/9 (10) | Neurodevelopmental delay | No | No | c.407G>A p.Cys136Tyr de novo |
| F16 | P20 | F | 2.9 months | 32 | 144 | Yes | Yes | Yes | Yes/13 (30) | Neurodevelopmental delay, dysmorphy | No | Short stature (final height of 1.48 m), obesity | c.407G>A p.Cys136Tyr nd |
| F17 | P21 | M | 9 months | 43 | 106 | Yes | Yes | Yes | Yes/5 (5) | Neurodevelopmental delay, extreme delayed speech | Yes (Lipomatosis) | IUGR | c.668C>A p.Ala223Asp de novo |
| F18 | P22 | M | 15 days | 1.5 (HSCT at 0.5) | 220 | Yes | N | Yes | Yes/26 (50) | Neurodevelopmental delay, epilepsy | No) | No | c.677G>A p.Gly226Glu de novo |
| F19 | P23 | M | 1 months | 25.6 | 92 | Yes | Yes | Yes | Yes/14 (30) | Neurodevelopmental delay, extreme delayed speech | Yes (Lipomatosis) | IUGR, short stature (final height of 1.53 m), bone dysplasia | c.821G>A p.Gly274Asp de novo |
| Family | ID | Sex | Age at diagnosis | Age at last follow-up (years) | ANCs (per microliter) before G-CSF therapy | Severe bacterial infections* | Stomatitis gingivitis | Maturation arrest on bone marrow smear (before G-CSF therapy) | G-CSF therapy mean (maximum) dose (µg/kg/day) | Neurological symptoms | Exocrine pancreatic insufficiency† | Other extrahematopoietic symptoms | SRP54 genotype cDNA; protein inheritance |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| F1 | P1 | M | 9.8 months | 8 | 270 | Yes | Yes | Yes | Yes/5 (5) | No | No | No | c.337G>C p.Gly113Arg de novo |
| F2 | P2 | M | 4.6 months | 3 | 443 | Yes | No | Yes | Yes/16 (20) | No | Yes | No | c.349_351del p.Thr117del de novo |
| F3 | P3 | F | 1.7 months | 15 | 440 | Yes | Yes | Yes | Yes/4 (5) | No | No | No | c.349_351del p.Thr117del dominant |
| P4 Father | M | 2.1 years | 44 | 100 | No | Yes | Yes | No | No | No | No | ||
| F4 | P5 | M | 10.6 months | 11 (HSCT at 1.5) | 190 | Yes | No | Yes | Yes/refractory till 50 µg/kg | Intellectual disability, autism spectrum disorder‡ | No | No | c.349_351del p.Thr117del nd |
| F5 | P6 | M | 5 years | 32 | 530 | No | Yes | Yes | Yes/16 (20) | No | No | Osteoporosis Type 2 diabetes at 31 years | c.349_351del p.Thr117del de novo§ |
| F6 | P7 | M | 2 days | 10 | 45 | Yes | Yes | Yes | Yes/6 (15) | No | No | No | c.349_351del p.Thr117del de novo |
| F7 | P8 | F | 10.1 months | 15 | 120 | No | Yes | Yes | Yes/10 (10) | No | No | No | c.349_351del p.Thr117del de novo |
| F8 | P9 | M | 1 months | 24 | 250 | Yes | Yes | Yes | Yes/5 (5) | No | No | IUGR, GH deficiency and GH therapy (Final height of 1.69 m) | c.349_351del p.Thr117del nd |
| F9 | P10 | M | 4.2 months | 11 | 230 | Yes | No | Yes | Yes/10 (10) | No | No | No | c.349_351del p.Thr117del de novo |
| F10 | P11‖ | M | 12.7 months | 7 | 360 | Yes | Yes | Yes | Yes/5 (5) | No | No | No | c.349_351del p.Thr117del de novo |
| F11 | P12‖ | M | 1.3 years | 39 | 90 | Yes | Yes | Yes | Yes/2 (5) | No | No | No | c.349_351del p.Thr117del de novo |
| F12 | P13‖ | F | 1 days | 3 | 100 | Yes | N | Yes | Yes/20 (20) | No | No | No | c.349_351del p.Thr117del de novo |
| F13 | P14 | M | 10 days | 6 | 110 | Yes | Yes | Yes | Yes/5 (5) | No | No | No | c.349_351del p.Thr117del dominant |
| P15 | F | 20 years | 28 | 345 | No | Yes | Yes | No | No | No | No | ||
| F14 | P16‖ | F | 4.2 months | 17 | 435 | Yes | No | Yes (intermittent) | Yes/5 (5) | No | No | No | c.353G>A p.Cys118Tyr dominant |
| P17‖ Father | M | 2.8 months | 46 | 960 | Yes | Yes | Yes (intermittent) | Yes/5 (5) | Learning difficulties** | No | No | ||
| P18 Brother | M | 8.3 months | 21 | 1090 | No | No | No | No | Learning difficulties** | No | No | ||
| F15 | P19‖ | F | 24 days | 6 | 215 | Yes | Yes | Yes | Yes/9 (10) | Neurodevelopmental delay | No | No | c.407G>A p.Cys136Tyr de novo |
| F16 | P20 | F | 2.9 months | 32 | 144 | Yes | Yes | Yes | Yes/13 (30) | Neurodevelopmental delay, dysmorphy | No | Short stature (final height of 1.48 m), obesity | c.407G>A p.Cys136Tyr nd |
| F17 | P21 | M | 9 months | 43 | 106 | Yes | Yes | Yes | Yes/5 (5) | Neurodevelopmental delay, extreme delayed speech | Yes (Lipomatosis) | IUGR | c.668C>A p.Ala223Asp de novo |
| F18 | P22 | M | 15 days | 1.5 (HSCT at 0.5) | 220 | Yes | N | Yes | Yes/26 (50) | Neurodevelopmental delay, epilepsy | No) | No | c.677G>A p.Gly226Glu de novo |
| F19 | P23 | M | 1 months | 25.6 | 92 | Yes | Yes | Yes | Yes/14 (30) | Neurodevelopmental delay, extreme delayed speech | Yes (Lipomatosis) | IUGR, short stature (final height of 1.53 m), bone dysplasia | c.821G>A p.Gly274Asp de novo |
ANC, absolute neutrophil count; F, female; GH, growth hormone; HSCT, hematopoietic stem cell transplant; IUGR, intrauterine growth retardation; M, male; nd, not determined.
Septicemia, pneumonia, cellulitis, liver abscess, and osteitis were considered as severe infections.
Exocrine pancreatic insufficiency requiring pancreatic enzyme therapy.
Father with severe neuropsychiatric disorder.
SRP54 mutation identified in the asymptomatic father at a mosaic state (9%; supplemental Figure 1).
Cases analyzed by WES.
Learning difficulties defined as requiring attendance at a special school.
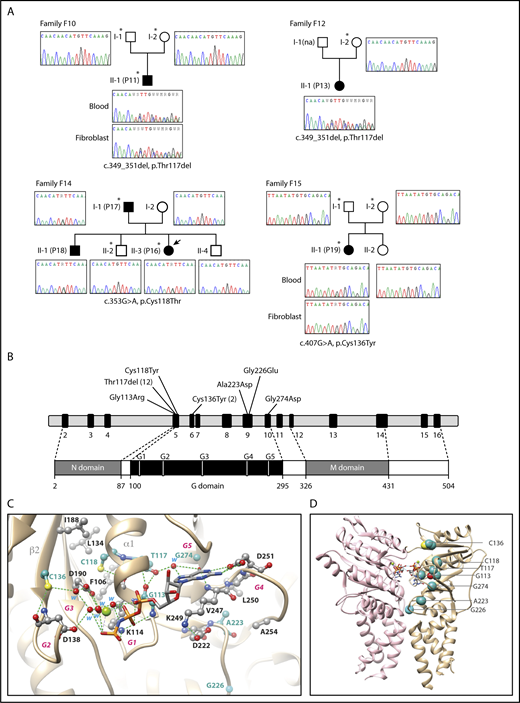
WES of 4 pedigrees and description of SRP54 mutations. (A) Confirmation of SRP54 mutations by Sanger sequencing of DNA extracted from whole blood sample and fibroblasts. *Individuals analyzed by WES. (B) Exon-intron of the SRP54 gene locus and protein diagram with the positions of all the mutations identified in this study. (C) Ribbon representation of the three-dimensional structure of the human SRP54 (gold)/SRα (pink) dimer (pdb 5l3q) in 2 different orientations, with the positions of the 7 mutated amino acids (sphere and ball-and-stick representations on the left and right, respectively). The 2 molecules of GMPPNP (5′-guanylyl-imidodiphosphate), a nonhydrolyzable GTP analog are shown. The positions of the 5 G elements are reported in green (G1) and red (G2-G5). Magnesium ions are shown as green balls. For a detailed description of the contacts and interactions made by the 7 mutated amino acids, see supplemental Data.
WES of 4 pedigrees and description of SRP54 mutations. (A) Confirmation of SRP54 mutations by Sanger sequencing of DNA extracted from whole blood sample and fibroblasts. *Individuals analyzed by WES. (B) Exon-intron of the SRP54 gene locus and protein diagram with the positions of all the mutations identified in this study. (C) Ribbon representation of the three-dimensional structure of the human SRP54 (gold)/SRα (pink) dimer (pdb 5l3q) in 2 different orientations, with the positions of the 7 mutated amino acids (sphere and ball-and-stick representations on the left and right, respectively). The 2 molecules of GMPPNP (5′-guanylyl-imidodiphosphate), a nonhydrolyzable GTP analog are shown. The positions of the 5 G elements are reported in green (G1) and red (G2-G5). Magnesium ions are shown as green balls. For a detailed description of the contacts and interactions made by the 7 mutated amino acids, see supplemental Data.
We subsequently sequenced SRP54 in 66 probands from the French CN Registry presenting a severe neutropenia diagnosed in childhood associated with a promyelocytic maturation arrest, and requiring G-CSF therapy for most of them. In all these patients, the most common genes involved in CN were previously excluded by next-generation sequencing of a 16-gene panel (supplemental Table 3). SRP54 mutations were identified in 15 of 66 probands, and then in 2 affected relatives (Table 1).
In total, SRP54 mutations were found in 23 cases (19 probands, 4 relatives). Most of them were sporadic cases (16/19). Parental DNA analysis confirmed the de novo occurrence of SRP54 mutations in the 13 tested cases. A paternal mosaicism detectable by Sanger sequencing was confirmed by high-throughput sequencing (Family F5, supplemental Figure 1).
Seven different SRP54 mutations were identified in the 19 probands (Table 1; Figure 1B). The in-frame deletion p.Thr117del was detected in 10 sporadic cases and in 2 families. The other variants were missense mutations (p.Gly113Arg, p.Cys118Tyr, p.Cys136Tyr, p.Ala223Asp, p.Gly226Glu, and p.Gly274Asp). These variants are all located in the central G domain that is crucial for the GTPase activity of SRP54 (Figure 1B) and were considered as likely pathogenic mutations (supplemental Table 4).20
Predictions of SRP54 mutation effect on the GDP/GTP activity and on the interaction with SRα receptor
We analyzed the experimental 3D structure of SRP54 to understand the possible effect of mutations. All the mutated amino acids occupy positions that are strictly or highly conserved within the SRP54 family, corresponding to small residues (Gly, Ala, Thr, Cys) and located for most of them within or in the vicinity of conserved G elements (Figure 1B-C; supplemental Figure 2). Any substitution with amino acids possessing larger side chains is predicted to lead to steric hindrance, affecting β-phosphate binding (p.G113 [G1 element]), GTP guanine binding (p.G274 [G5 element]), magnesium coordination through water molecules (p.Cys136), or on a more global scale, the stability of the GTP-binding domain structure (p.Cys118 and p.Ala223) or SRP54/SRα interaction (p.Gly226; for a detailed description, see supplemental Data). Deletion of p.Thr117 is predicted to affect the three-dimensional structure or stability of the core α1 helix preceding the P-loop (G1 motif), thereby possibly affecting GTP binding through modification of the P-loop conformation.
Genotype-phenotype relationships define 2 groups of patients
The patients presented a chronic and severe neutropenia with a median absolute neutrophil count of 0.22 × 109/L that was diagnosed in the neonate period or during childhood (median age, 4.2 months) because of the occurrence of severe bacterial infections in the majority of individuals (18/23, 78%; Table 1). Elevated circulating monocyte counts (1.37 × 109/L) were observed and frequently associated with elevated lymphocyte counts (3.9 × 109/L; supplemental Table 5). During routine follow-up, 7 individuals had transient moderate anemia (hemoglobin <9 g/dL) and 2 had a transient thrombocytopenia with platelet count below 100 × 109/L (supplemental Table 5).
Nearly all patients (20/23, 87%) required G-CSF therapy (Table 1). The optimal G-CSF doses were between 5 and 10 μg/kg/day in 9 cases (45%), between 10 and 30 μg/kg/day in 7 patients (35%), and above 30 μg/kg/day in 4 patients (20%). Two patients (P5, P22) were considered fully refractory after G-CSF therapy above 50 μg/kg/day and received allogeneic hematopoietic stem cell transplantation (Table 1). Despite high doses of G-CSF, no evolution into acute myeloid leukemia (AML) was observed after a median follow-up period of 14.8 years with a range between 0.5-46 years. The targeted high-density resequencing of CSF3R, RUNX1, and TP53 genes, mutated in the CN evolving to AML,21-23 did not reveal any acquired events on DNA extracted from either bone marrow (n = 6) or whole blood samples (n = 11) of 17 patients (data not shown). Of note, 16/18 individuals with SRP54 mutations interacting directly with the G1 element were associated with only a hematological phenotype (Table 1; Figure 1B-C). The 2 exceptions were patients P2 and P5. Patient P2 presented a moderate exocrine pancreatic insufficiency at the age of 3 years. Patient P5 had a severe intellectual disability with autistic traits. As he had a familial history of severe neurological disorders without known neutropenia, we could not exclude that his neurodevelopmental delay is a result of another cause.
In contrast, the 5 other patients with SRP54 mutations located within or in the vicinity of G2, G4, and G5 elements (Figure 1B-C; supplemental Data) were associated with a neurodevelopmental delay characterized by extreme delayed speech and intellectual disability. Those SRP54 mutations (Cys136Tyr, Ala223Asp, Gly226Glu, and Gly274Asp) are predicted not to be involved in GTP phosphate binding or hydrolysis, but rather in more structural issues such as guanine binding or SRP54/SRα interaction. Exocrine pancreatic deficiency requiring pancreatic enzyme supplementation was present in 2 of them (P21 and P23; Table 1). In contrast, only 1 case among the 18 patients with mutations located within the G1 element had exocrine pancreatic deficiency. However, subclinical pancreatic dysfunction was suggested by the reduced level of fat-soluble vitamin A and pancreatic enzymes (fecal elastase or serum lipase) in 53% and 27% of these patients, respectively (supplemental Table 6).
The morphological aspects of granulocytic cells are characteristic of the SRP54-mutated entity
Analysis of bone marrow smears showed a promyelocytic maturation arrest with a significant decrease in the percentage of myeloid precursors and neutrophils in all individuals except 1 (P18). Patient P18 belongs to an autosomal-dominant family (F14) with 2 other individuals presenting an intermittent maturation defect.
Cytological analysis of 18 patients revealed a high degree of dysgranulopoiesis (25%-80%). Promyelocytes had numerous large cytoplasmic vacuoles, condensed granulations located in the Golgi (Figure 2Aa-b). Some promyelocytes clearly displayed apoptotic characteristics (Figure 2Ac-d), abnormal mitosis (Figure 2Ae), and double nuclei (Figure 2Af). The remaining neutrophil granulocytes were dystrophic with double nuclei and abnormal clumped chromatin (Figure 2B). In contrast to patients with ELANE mutations, dysgranulopoiesis was systematically present in SRP54-mutated cases and with a higher number of cells with dysgranulopoietic features (P = .02; Figure 2C). Transmission electron microscopy (TEM) of the bone marrow of patient P1 (Figure 2D-E; supplemental Figure 3) showed dystrophic promyelocytes with enlarged ER located around the nucleus or throughout the cytoplasm surrounded by numerous ribosomes. Cytoplasm also contains few autophagosomes (Figure 2D). Analysis of nuclei confirmed the apoptotic features of myeloid cells and neutrophils (Figure 2E).

Cytological and ultrastructural characteristics of bone marrow granulocytic cells. (A-B) Cytology analysis after May-Grünwald Giemsa staining. Pictures represent granulocytic precursors (a-f) and neutrophils (g-i)) in different individuals P3, P10, P19, P20, and P23. (Original magnification ×100). (C) Comparative cytological bone marrow analysis of 16 individuals with SRP54 mutations and 16 patients with ELANE mutations Significant difference was observed between SRP54 and ELANE patients using nonparametric Kruskal-Wallis test (P = .02). (D-E) Analysis by TEM of the ultrastructural aspect of bone marrow cells from the granulocytic lineage in patient P1. Enlarged ER was observed around the nucleus (N) and thorough the cytoplasm (Cyt) in promyelocytes (D). The black arrow points to nascent autophagosome. Right panels (E) show pre-apoptotic neutrophil granulocytes, a major characteristic of the sample.
Cytological and ultrastructural characteristics of bone marrow granulocytic cells. (A-B) Cytology analysis after May-Grünwald Giemsa staining. Pictures represent granulocytic precursors (a-f) and neutrophils (g-i)) in different individuals P3, P10, P19, P20, and P23. (Original magnification ×100). (C) Comparative cytological bone marrow analysis of 16 individuals with SRP54 mutations and 16 patients with ELANE mutations Significant difference was observed between SRP54 and ELANE patients using nonparametric Kruskal-Wallis test (P = .02). (D-E) Analysis by TEM of the ultrastructural aspect of bone marrow cells from the granulocytic lineage in patient P1. Enlarged ER was observed around the nucleus (N) and thorough the cytoplasm (Cyt) in promyelocytes (D). The black arrow points to nascent autophagosome. Right panels (E) show pre-apoptotic neutrophil granulocytes, a major characteristic of the sample.
Increased expression of SRP54 during granulocyte differentiation
To decipher the role of SRP54 in the granulocytic differentiation, we cultured normal CD34+ progenitors in vitro in serum-free medium in the presence of cytokines (SCF+IL-3+G-CSF) for 21 days. Cytology analysis showed at day 7 a majority of immature cells (myeloblasts and promyelocytes), at day 14 a mix of immature and mature cells (promyelocytes, metamyelocytes, myelocytes, immature monocytes/monoblasts), and at day 21 an enrichment in mature cells (neutrophils and monocytes; Figure 3A). During differentiation, we quantified the SRP54 expression levels and observed a 6-fold increase that coincided with an increase in CSF3R expression. No significant induction was found for SRP72, another protein of the SRP complex previously identified as a predisposition gene to familial aplastic anemia (Figure 3B-D).24 In contrast, SRP54 expression remained unchanged from progenitors (CD34+) to megakaryocytes (CD41+CD42+) and erythroblasts (CD36+GPA+), suggesting a specific role in the granulocytic cells (Figure 3E). SRP54 mRNA and protein expression levels were not significantly different between controls and patients (Figure 3F-G), suggesting a protein function defect rather than a quantitative defect.
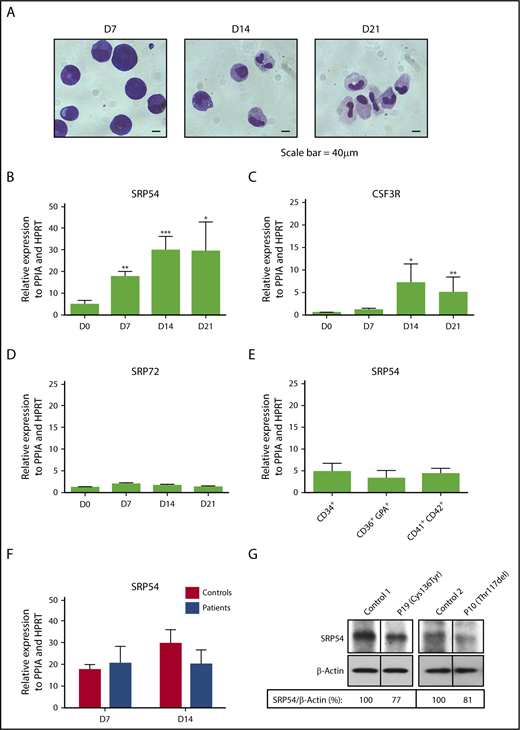
Increased expression of SRP54 during granulocyte differentiation. CD34+ cells from healthy donors (controls) or patients were cultured for 21 days in serum free-medium with SCF, IL-3, and G-CSF. (A) May-Grünwald Giemsa was performed to determine the purity and stages of differentiation at days (D) 7, 14, and 21. The expression levels of (B) SRP54, (C) CSF3R, and (D) SRP72 were quantified by quantitative reverse transcription polymerase chain reaction (qRT-PCR) related to PPIA and HPRT at days (D) 0, 7, 14, 21. Results are the mean ± SEM of 4 to 8 control samples performed in triplicate. Student t test: *P < .05; **P < .01; ***P < .001. (E) The expression level of SRP54 was quantified by qRT-PCR related to PPIA and HPRT in CD34+ progenitors, CD36+GPA+ erythroblasts, and CD41+CD42+ megakaryocytes. Results are the mean ± SEM of 4 to 8 control samples performed in triplicate. (F) The expression level of SRP54 was quantified by qRT-PCR related to PPIA and HPRT in granulocytic precursors at days 7 and 14 in both controls (n = 5) and patients (n = 5). Results are the mean ± SEM performed in triplicate. (G) The expression level of SRP54 protein was evaluated by western blot analysis on day 10 granulocytic precursors in controls (n = 2) and patients (n = 2), using specific antibodies.
Increased expression of SRP54 during granulocyte differentiation. CD34+ cells from healthy donors (controls) or patients were cultured for 21 days in serum free-medium with SCF, IL-3, and G-CSF. (A) May-Grünwald Giemsa was performed to determine the purity and stages of differentiation at days (D) 7, 14, and 21. The expression levels of (B) SRP54, (C) CSF3R, and (D) SRP72 were quantified by quantitative reverse transcription polymerase chain reaction (qRT-PCR) related to PPIA and HPRT at days (D) 0, 7, 14, 21. Results are the mean ± SEM of 4 to 8 control samples performed in triplicate. Student t test: *P < .05; **P < .01; ***P < .001. (E) The expression level of SRP54 was quantified by qRT-PCR related to PPIA and HPRT in CD34+ progenitors, CD36+GPA+ erythroblasts, and CD41+CD42+ megakaryocytes. Results are the mean ± SEM of 4 to 8 control samples performed in triplicate. (F) The expression level of SRP54 was quantified by qRT-PCR related to PPIA and HPRT in granulocytic precursors at days 7 and 14 in both controls (n = 5) and patients (n = 5). Results are the mean ± SEM performed in triplicate. (G) The expression level of SRP54 protein was evaluated by western blot analysis on day 10 granulocytic precursors in controls (n = 2) and patients (n = 2), using specific antibodies.
We also investigated the effects of the SRP54 mutations on the granulocytic differentiation and found a 10-fold decrease in patient cell proliferation after 14 days compared with control cells (Figure 4A-B; supplemental Figure 4). Of note, there was no correlation between the type of mutations and the inhibition of cell proliferation. This was associated with a 2-fold increase in apoptotic cells in SRP54-mutated patient cells compared with controls, using the annexin-V/propidium iodide assay (Figure 4C-D; supplemental Figure 5). Moreover, we found an enrichment in P53 target genes specifically related to apoptosis (BAX, NOXA1, P21) in patient cells compared with control patients. These results were confirmed in sorted mature and immature granulocytic cells (CD36−CD15+CD11b− and CD36−CD15+CD11b+; Figure 4E-F). In addition, we found an impaired signaling of day 10 granulocytic cells in response to various doses of G-CSF (ie, increased STAT3 phosphorylation and concomitant decreased in AKT and ERK phosphorylation in patient cells compared with controls; supplemental Figure 4).
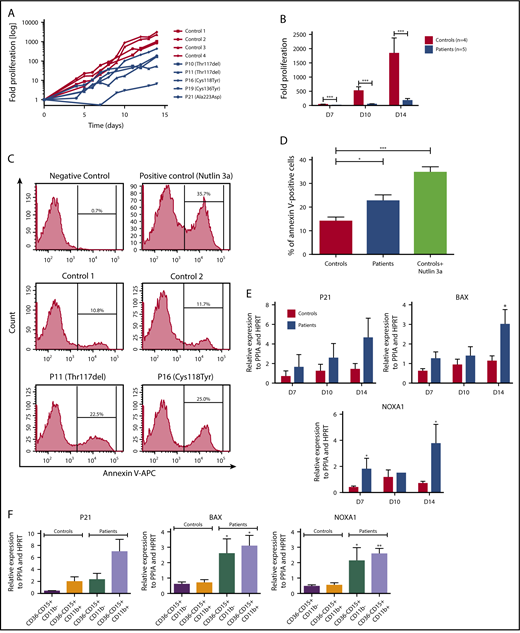
SRP54 mutations induce a great impairment of granulocytic differentiation. CD34+ cells from healthy donors (controls) or patients were cultured for 14-21 days in serum free-medium with SCF, IL-3, and G-CSF. (A) The fold increase in proliferation in log scale was assessed individually by Trypan blue exclusion, using controls (= 4) and patients (n = 5). (B) Fold increase in proliferation in patients and controls at days (D) 7, 10, and 14. Mean ± SEM; ***P < .001. (C) Apoptotic granulocytic cells were analyzed by flow cytometry using Annexin V+ assay in controls (n = 2) and patients (n = 2). The MDM2 antagonist, Nutlin 3a (20 µM for 24 hours), was used as positive control at D9. (D) The results represent the mean of 3 independent experiments at days 7, 9, and 13 with controls (n = 2) and patients (n = 2). Mean ± SEM; *P < .05; ***P < .001. (E) P53 target genes (P21, BAX, NOXA) expression levels were checked by qRT-PCR related to PPIA and HPRT at days (D) 7, 10, and 14 with controls (n = 3-4) and patients (n = 2-4). Mean ± SEM; *P < .05. (F) Granulocytic cells were sorted as CD15+CD11b+CD36− cells between days 10 and 14, using CD36 to specifically eliminate monocytes. P53 target genes (P21, BAX, NOXA) expression levels were checked by qRT-PCR related to PPIA and HPRT with controls (n = 4) and patients (n = 3). Mean ± SEM; *P < .05; **P < .01.
SRP54 mutations induce a great impairment of granulocytic differentiation. CD34+ cells from healthy donors (controls) or patients were cultured for 14-21 days in serum free-medium with SCF, IL-3, and G-CSF. (A) The fold increase in proliferation in log scale was assessed individually by Trypan blue exclusion, using controls (= 4) and patients (n = 5). (B) Fold increase in proliferation in patients and controls at days (D) 7, 10, and 14. Mean ± SEM; ***P < .001. (C) Apoptotic granulocytic cells were analyzed by flow cytometry using Annexin V+ assay in controls (n = 2) and patients (n = 2). The MDM2 antagonist, Nutlin 3a (20 µM for 24 hours), was used as positive control at D9. (D) The results represent the mean of 3 independent experiments at days 7, 9, and 13 with controls (n = 2) and patients (n = 2). Mean ± SEM; *P < .05; ***P < .001. (E) P53 target genes (P21, BAX, NOXA) expression levels were checked by qRT-PCR related to PPIA and HPRT at days (D) 7, 10, and 14 with controls (n = 3-4) and patients (n = 2-4). Mean ± SEM; *P < .05. (F) Granulocytic cells were sorted as CD15+CD11b+CD36− cells between days 10 and 14, using CD36 to specifically eliminate monocytes. P53 target genes (P21, BAX, NOXA) expression levels were checked by qRT-PCR related to PPIA and HPRT with controls (n = 4) and patients (n = 3). Mean ± SEM; *P < .05; **P < .01.
SRP54 mutants lead to ER-stress and autophagy
To understand the SRP54 mutation-induced apoptosis, we investigated the presence of ER stress and autophagy, as suggested by TEM observations. Using in vitro-derived granulocytic cells in bulk or after cell sorting, we found that SRP54 mutants induce a significant increase in ATF4, CHOP, and spliced XBP1 expression levels, pointing out an ER stress (Figure 5A-C). ULK1 expression level was also significantly upregulated in primary cells (Figure 5B-D), and an increase in LC3-II was observed in SRP54-mutated primary fibroblasts, suggesting the induction of autophagy (Figure 5E). Moreover, the co-localization of SRP54 with calreticulin (CALR), an ER/Golgi marker, was scrutinized by confocal microscopy. Although in the normal fibroblasts, the CALR presents a classical localization of the ER/Golgi, CALR labeling was increased and aggregated in the SRP54-mutant fibroblasts (supplemental Figure 6).

SRP54 mutant induce ER-stress and autophagy. mRNA expression levels of (A) ATF4, CHOP, and spliced XBP1 for ER stress and (B) ULK1 for autophagy were examined by qRT-PCR related to PPIA and HPRT at days (D) 7, 10, and 14 of culture of control (n = 3-4) and patient (n = 2-4) cells. Mean ± SEM; unpaired t test, 2-tailed on controls vs patients combing all days, *P < .05. mRNA expression levels of (C) CHOP and (D) ULK1 were also checked after sorting CD36−CD15+CD11b− and CD15+CD11b+CD36− granulocytic cells between days 10 and 14. Controls (n = 3) and patients (n = 3); mean ± SEM; unpaired t test, 2-tailed on controls vs patients; *P < .05; **P < .01. (E) Using primary fibroblasts, autophagy was evaluated by the lipidation of LC3 (LC3-II) by western blot analysis in controls (n = 2) and SRP54-mutated patients (n = 8). β-Actin was used as a loading control. Fold induction of LC3-II/β-Actin was quantified using Image J software.
SRP54 mutant induce ER-stress and autophagy. mRNA expression levels of (A) ATF4, CHOP, and spliced XBP1 for ER stress and (B) ULK1 for autophagy were examined by qRT-PCR related to PPIA and HPRT at days (D) 7, 10, and 14 of culture of control (n = 3-4) and patient (n = 2-4) cells. Mean ± SEM; unpaired t test, 2-tailed on controls vs patients combing all days, *P < .05. mRNA expression levels of (C) CHOP and (D) ULK1 were also checked after sorting CD36−CD15+CD11b− and CD15+CD11b+CD36− granulocytic cells between days 10 and 14. Controls (n = 3) and patients (n = 3); mean ± SEM; unpaired t test, 2-tailed on controls vs patients; *P < .05; **P < .01. (E) Using primary fibroblasts, autophagy was evaluated by the lipidation of LC3 (LC3-II) by western blot analysis in controls (n = 2) and SRP54-mutated patients (n = 8). β-Actin was used as a loading control. Fold induction of LC3-II/β-Actin was quantified using Image J software.
SRP54 knockdown mimics the functional defect, including decreased proliferation and increased ER stress observed in granulocytic cells of SRP54 individuals
To understand whether the SRP54 mutations act as gain- or loss-of-function, we generated 3 lentiviruses expressing either shRNA targeting SRP54 or a scramble sequence (SCR) and GFP as a selection marker and transduced the HL-60 cell line. The 2 specific shRNA inhibited the SRP54 transcript by 50% (Figure 6A). Meanwhile, SRP54 knockdown induced a slower proliferation of HL-60 cells (Figure 6B). As previously observed in SRP54 mutant cells, SRP54 knockdown led not only to an augmentation of spliced XBP1 mRNA expression, and the phosphorylation of both eIF2a and PERK, showing an ER stress, but also to an increased phosphorylation of ULK1, a marker of autophagy (Figure 6C-D). These experiments were also performed in primary cells from healthy donors after transduction with the shRNA, and subsequent GFP+-sorting of in vitro cultured cells. In CD36−CD15+CD11b+ cells, we found that only shRNA_2 was efficient and led to decreased cell proliferation with a trend in increased spliced XBP1 (Figure 6E-G).

SRP54 knockdown induces a defect in proliferation and increased ER stress and autophagy. (A-D) HL-60 cell lines stably expressing either shRNA targeting SRP54 (1 or 2) or a SCR were generated after transduction with lentiviral particles and after sorting on GFP+ after 72 hours. Nontransduced (NT, not transduced) HL-60 cell line was used as control. (A) SRP54 mRNA expression level was checked by qRT-PCR related to PPIA (n = 4). Mean ± SEM; unpaired t test, 2-tailed; *P < .05; **P < .01. (B) Fold proliferation was assessed after Trypan blue exclusion for 3 days (n = 4). Mean ± SEM; unpaired t test, 2-tailed; *P < .05. (C) Spliced XBP1 mRNA expression level was checked by qRT-PCR related to PPIA, CFL1, and H2AZ (n = 4). Mean of SCR or shRNA_1 and 2 ± SEM; Unpaired t test, 2-tailed; *P < .05. (D) P-eIF2a, P-PERK, P-ULK1 were evaluated by western blot analysis using specific antibodies. β-Actin was used as loading control. (E-G) Normal CD34+ progenitors were transduced with 2 lentiviruses expressing either shRNA targeting SRP54 (ShSRP54_2) or SCR. CD34+GFP+ cells were sorted 72 hours later and were cultured for 10 days in serum-free medium with SCF, IL-3, and G-CSF. (E) Granulocytic cells were counted by Trypan blue exclusion (n = 3) or sorted the same day on the CD36−CD15+CD11b+ phenotype for analysis of (F) SRP54 gene expression level (n = 4) and (G) spliced XBP1 (n = 3) mRNA expression level by qRT-PCR related to PPIA and HPRT. Mean ± SEM; unpaired t test, 2-tailed; **P < .01; ***P < .001.
SRP54 knockdown induces a defect in proliferation and increased ER stress and autophagy. (A-D) HL-60 cell lines stably expressing either shRNA targeting SRP54 (1 or 2) or a SCR were generated after transduction with lentiviral particles and after sorting on GFP+ after 72 hours. Nontransduced (NT, not transduced) HL-60 cell line was used as control. (A) SRP54 mRNA expression level was checked by qRT-PCR related to PPIA (n = 4). Mean ± SEM; unpaired t test, 2-tailed; *P < .05; **P < .01. (B) Fold proliferation was assessed after Trypan blue exclusion for 3 days (n = 4). Mean ± SEM; unpaired t test, 2-tailed; *P < .05. (C) Spliced XBP1 mRNA expression level was checked by qRT-PCR related to PPIA, CFL1, and H2AZ (n = 4). Mean of SCR or shRNA_1 and 2 ± SEM; Unpaired t test, 2-tailed; *P < .05. (D) P-eIF2a, P-PERK, P-ULK1 were evaluated by western blot analysis using specific antibodies. β-Actin was used as loading control. (E-G) Normal CD34+ progenitors were transduced with 2 lentiviruses expressing either shRNA targeting SRP54 (ShSRP54_2) or SCR. CD34+GFP+ cells were sorted 72 hours later and were cultured for 10 days in serum-free medium with SCF, IL-3, and G-CSF. (E) Granulocytic cells were counted by Trypan blue exclusion (n = 3) or sorted the same day on the CD36−CD15+CD11b+ phenotype for analysis of (F) SRP54 gene expression level (n = 4) and (G) spliced XBP1 (n = 3) mRNA expression level by qRT-PCR related to PPIA and HPRT. Mean ± SEM; unpaired t test, 2-tailed; **P < .01; ***P < .001.
Discussion
Here, we describe a novel genetic subtype of CN associated with de novo or dominantly inherited mutations in SRP54 gene. This genetic entity represents the second most common cause of CN with maturation arrest in the French CN registry with a prevalence of 6.9%. All patients with SRP54 mutations were characterized by a chronic and profound neutropenia occurring within the first months of life and a maturation arrest at the promyelocytic stage. The hematological profile of patients with SRP54 mutations was the same as the one observed in patients with ELANE mutations.25 However, the analysis of bone marrow cells showed a major and constant dysgranulopoiesis differentiating SRP54 patients from ELANE patients who displayed a lower degree of dysgranulopoiesis. Similarly, to patients with CSF3R or JAGN1 deficiency, but in contrast to patients with ELANE mutations, nearly all patients with SRP54 mutations showed a poor or an absence of response to G-CSF therapy. Interestingly, despite high doses of G-CSF, none of the patients in the cohort transformed to leukemia after a median follow-up of 15 years. In contrast, a high incidence of AML occurred in ELANE-mutated patients with CN treated by G-CSF as a result of clonal expansion of CSF3R-mutated cells.21,23,26 The absence of acquired CSFR3 mutations in SRP54 patients may be related to the low response to G-CSF therapy, suggesting that in this context, CSFR3 mutations cannot be selected.
SRP54 encodes for a GTPase, which is the core component of the cytosolic ribonucleoprotein complex, a universally conserved system that mediates the cotranslational targeting of newly synthesized proteins emerging from the ribosome and destined for secretion or ER membrane insertion by binding their N-terminal signal sequence.14,19 Subsequently, the ribonucleoprotein complex bound to the ribosome-nascent chain complex is targeted to the ER by GTP-dependent interaction between SRP54 and the membrane-bound SRP54 receptor. This ER targeting regulates the entire cotranslational process.15 In human, the ribonucleoprotein complex consists of 6 proteins (SRP9, SRP14, SRP19, SRP54, SRP68, and SRP72) and a single 7S RNA.19 Apart from SRP54, mutations in the SRP72 gene have been described in 2 families with autosomal dominant form of aplastic anemia and myelodysplasia.24 One should note that 2 young family members, aged 11 and 12 years old and carriers of the SRP72 mutation, were characterized by a neutropenia and a moderate thrombocytopenia.
SRP54 has 3 functional domains, the N-terminal domain (N-domain) tightly connected to the central GTPase domain (G domain) implicated in the GTP-dependent interaction with SR and forming the functional unit named the NG domain, and the M domain, involved in the signal sequence binding. The G domain contains several specific G elements, G1 to G5, involved in nucleotide binding and hydrolysis. The 7 different SRP54 mutations identified in our study were all located in the G domain and predicted to affect either the catalytic function of SRP54 or the 3D structure of the NG domain implicated in GTP binding and dimerization with the SR receptor. In accordance with these data, Carapito et al showed a global decrease in the GTPase activity of the SRP54 mutants, and in particular with the Gly226Glu mutation.27 Our study suggests genotype-phenotype relationships. Indeed, except for 2 cases, the 18 patients with SRP54 mutations located within the G1 element had only a hematological phenotype. The other patients with mutations interfering with G4 or G5 presented a severe neurodevelopmental disorder, and for some of them, an exocrine pancreatic insufficiency. Carapito et al reported heterozygous SRP54 mutations in 3 sporadic cases with neutropenia and Shwachman-Diamond (SDS)-like features.27 Among them, the sole individual with developmental delay and exocrine pancreatic deficiency was associated with the Gly226Glu mutation located outside the G1 element, which is the most functionally deleterious.
Even if some individuals with SRP54 mutations presented extrahematopoietic manifestations classically associated with SDS, several aspects differentiate them. Indeed, in patients with SDS, neutropenia is generally moderate and associated with a promyelocytic arrest in only about one-fourth of cases, with dysgranulopoietic features being only observed at late stages of granulocytic differentiation28 ; anemia is reported in up to 80% and thrombocytopenia between 24% to 88% of patients with SDS29 ; a clonal myeloid disease with acquired chromosomal characteristics leading to myelodysplastic syndrome and AML28,30 is a complication of SDS; and metaphyseal dysplasia, skin, or cardiac defects are frequent.
Compared with hematopoietic progenitors, erythroblasts, and megakaryocytes, SRP54 is predominantly expressed in granulocytic cells, particularly during late stages of differentiation, from promyelocytes to neutrophils. Promyelocytes are characterized by the development of primary granules containing an abundant amount of proteases that are processed in the ER.31,32 As a consequence, these proteins require the ribonucleoprotein complex cellular machinery to follow the secretory pathway.
We showed that SRP54 mutations, independent of the location in the G domain, led to a drastically reduced proliferation of granulocytic cells. The proliferation defect was associated with an increase of apoptosis, a mechanism common to other genetic subtypes of CN.3 Increased expression of BAX1 and NOXA1 genes suggested that apoptosis in SRP54-CN was dependent on the p53 pathway, as observed in SDS.23,33 and other inherited bone marrow failure syndromes such as Diamond-Blackfan anemia.3,34
The SRP54 mRNA and protein expression was unchanged in neutrophils of patients compared with control patients. This result contrasts with the significant decreased SRP54 expression recently reported on whole bone marrow cells of patients.27 As normal bone marrow cells are composed of 60% to 70% granulocytic cells, this abnormal expression level may simply reflect the decrease in myeloid precursors in patient bone marrows. Consistent with a SRP54 loss-of-function defect, its knockdown led to a decreased proliferation. Accordingly, SRP54 was shown to be essential in yeast, and depletion or mutated forms of SRP54 that alter the GTPase activity induced lethality or conditional phenotype.35 Moreover, the srp54-knockdown zebrafish model showed a decreased number of neutrophils and reduced chemotaxis.27
Cytological and TEM-based analyses of bone marrow smears showed evidence of a major dysgranulopoiesis and suggested that SRP54 mutations may lead to cellular ER stress and autophagy. This was confirmed by SRP54 knockdown in the HL60 cell line and in primary cells. The SRP54 defect may lead to ER stress and subsequent autophagy through abnormal targeting and translocation of some secretory proteins involving essential proteins for granulocytic differentiation or essential ER/Golgi resident proteins for maturation of secretory or membrane-embedded proteins. Indeed, BIP, CALR, and alkaline phosphatase were previously found downregulated by SRP54 knockdown that may result in increase of unfolded proteins responsible for the ER stress.36 In agreement, the G-CSF receptor could be mislocalized in SRP54-mutated context as we observed a decreased phosphorylation of AKT and ERK on G-CSF stimulation, suggesting a reduced cell surface signaling.37 Our data underline the role of ER stress, which is a common feature of CN.3,4 In addition, a direct activation of autophagy may be induced by defective SRP54 through the elimination of endogenous defective proteins by UBQLN4.38,39 Both induction of ER stress and autophagy overload may explain the apoptosis of SRP54 mutated cells, but we do not know at present whether it is a result of a direct P53 activation. Similar mechanisms could also be involved in pancreatic cells, which secrete a large amount of enzymes.40
In conclusion, we characterized a pathological pathway in CN implicating the universally conserved SRP54 protein that cotranslationally targets nascent secretory and membrane proteins to the ER. Loss-of-function mutations of SRP54 lead to profound neutropenia by apoptosis associated with ER stress and the induction of autophagy.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the families for their participation and collaboration; Béatrice Parfait and Patrick Nusbaum for providing fibroblast cultures (Center of Biological Resources, Hôpital Cochin, Paris, France); and Philippe Rameau, Yann Lecluse, Cyril Catelain, and Frédéric De Leeuw from “Plateforme d’Imagerie et de Cytométrie.”
This work was supported by grants from the Association pour la Recherche contre le Cancer (ARC), Fondation ARC libre 2012 and the Institut National du Cancer Plbio (2015, 2018 to I. Plo), from the Fondation Maladies Rares (2015), Centre de Référence des Déficits Immunitaires Héréditaires (2015 to C.B.-C.). The program “Investissements d’avenir” is founding the Labex GR-Ex (I. Plo, W.V.). B.S.-P. was funded by Molecular Medicine in Oncology, Agence Nationale de la Recherche (MMO, ANR) and then by the Association de la Recherche sur la Moelle Osseuse. The French registry is supported by grants from Chugai SA, Prolong Pharma SA and Inserm. This project is supported by constant and unlimited support of the Association Sportive de Saint Quentin Fallavier since 2004, with the unlimited commitment of Christian Gonnot. The authors thank the association Immuno-déficience primitive, Recherche, Information, Soutien and Virginie Grosjean for her support.
Authorship
Contribution: C.B.-C., I. Plo, and J.D. designed the study, analyzed the data, and wrote the manuscript; C.B.-C. analyzed the exome sequencing; S.C. and A.D. performed and analyzed the genetic analysis; O.F. reviewed bone marrow cytology; I.C. performed the structural analysis; S.S. and G.P. performed and analyzed the transmission electronic microscopy; V.S. performed cytological analyses of cell cultures; B.S.-P., C.M., I.A.-D., and I. Plo performed the functional studies; G.-L.D., T.L., I. Pellier, C.S., N.A., V.B., F.B., P.B., O.B.-T., C.B., Y.B., L.C., C.F., C.G., A.S., H.J., F.M., B.N., T.C.Q., E.O., M.O., M.P., and F.S. provided samples and clinical data; B.B. collected the clinical data in the French Registry of Chronic Neutropenia; W.V. discussed the results; C.M. and W.V. reviewed and edited the manuscript; and all authors approved the final version of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Christine Bellanné-Chantelot, Département de Génétique, Hôpital Pitié-Salpêtrière, 47-83 Bd de l'Hôpital, 75651 Paris Cedex 13, France, UMR U1170, Gustave Roussy, 114 rue Edouard Vaillant, 94805 Villejuif, France; e-mail: christine.bellanne-chantelot@aphp.fr.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal