

TO THE EDITOR:
Inherited and de novo heterozygous germ line mutations in GATA2 cause a pleotropic autosomal dominant genetic disorder characterized by cellular deficiencies with a high propensity for myeloid malignancy.1,2 The clinical syndromes of GATA2 deficiency include monocytopenia and susceptibility to Mycobacterium avium complex infections (MonoMac); dendritic cell, monocyte, B, and natural killer lymphoid (DCML) deficiency3-6 ; sensorineuronal deafness and primary lymphedema with a predisposition for myelodysplastic syndrome (MDS) or acute myeloid leukemia (AML) (Emberger syndrome)7 ; familial MDS/AML8 ; pediatric MDS9 ; chronic neutropenia10 ; and bone marrow failure.11 GATA2 encodes a major zinc finger hematopoietic transcription factor that can occupy GATA-DNA motifs in several thousand genes.12 Its role in early hematopoiesis is achieved by cooperating in a complex network of transcription factors.13-15 Approximately 100 different mutant alleles have been identified that attenuate GATA2 transcriptional activity via deletion, chain termination, point mutations, or promoter/enhancer16 mutations that lead to decreased expression.17 Alternatively, GATA2 T354M may act in a dominant-negative fashion.8 We report here a novel, exonic synonymous mutation activating a cryptic splice donor site within GATA2 exon 3 that caused immunodeficiency in a single patient and inherited AML and MDS in a separate family. This work was approved by the institutional review boards of the University of Washington and University of Freiburg in compliance with the Declaration of Helsinki.
A 48-year-old woman presented with recurrent human papillomavirus–associated cervical intraepithelial neoplasia, extensive lower extremity deep vein thrombosis, chronic hepatitis C virus infection, and recurrent pneumonias complicated by cryptogenic organizing pneumonia and pulmonary alveolar proteinosis (Figure 1A). Laboratory findings revealed monocytopenia and B-cell, natural killer cell, dendritic cell, and CD4+ T-cell cytopenias (Figure 1A). Family history at presentation was uneventful for myeloid malignancies and immunodeficiency (additional clinical history in supplemental Data; available on the Blood Web site).
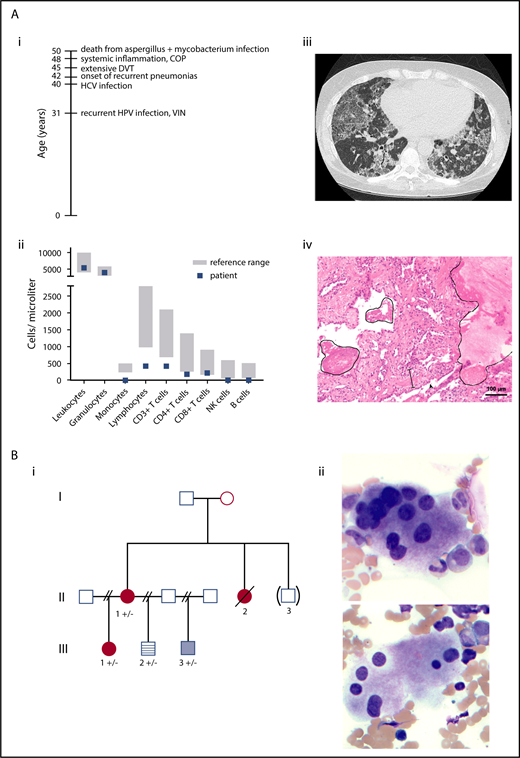
Clinical information on patients. (A) Immunodeficiency patient. (i) Timeline of complications. (ii) Leukocyte and lymphocyte subpopulations at 48 years old. (iii) Computed tomography scan of the lungs at 50 years old showed “crazy paving” as a dominant feature with diffusely distributed ground glass opacities and thickening of the interlobular septa. Additionally, bronchiectasis and reticulations were seen. All findings are compatible with a chronic interstitial lung disease seen in alveolar proteinosis. (iv) Lung biopsy sample with protein rich exudates (line), interstitial broadening with fibroblast proliferations and scattered lymphocytes (spacing line), and hyperplasia of type II pneumocytes (arrowhead). Patient age 50 years old. Hematoxylin and eosin stain, magnification ×10; Olympus BX-51 microscope and Zeiss AxioCamMR3 camera. (B) Inherited AML/MDS family. (i) Inherited AML/MDS pedigree. Circle, female; square, male; filled red circles, history of AML/MDS; pale blue box, history of thrombocytopenia; blue striped box, normal blood counts (no further workup); unfilled boxes, individuals reported to have normal blood counts. II-1 and III-2 had ptosis. “( )”, adopted-out of the family; “+”, GATA2 wild-type; “-”, p.Thr117= GATA2. (ii) Dysplastic megakaryocytes characterized as mononuclear and with separated nuclear lobes. Pre-PBSCT marrow aspirate from II-1. Wright-Giemsa stain, magnification ×40; Leica DM 2500 microscope and MC170 HD camera. COP, cryptic organizing pneumonia; DVT, deep vein thrombosis; HCV, hepatitis C virus; HPV, human papillomavirus; VIN, vaginal intraepithelial neoplasia.
Clinical information on patients. (A) Immunodeficiency patient. (i) Timeline of complications. (ii) Leukocyte and lymphocyte subpopulations at 48 years old. (iii) Computed tomography scan of the lungs at 50 years old showed “crazy paving” as a dominant feature with diffusely distributed ground glass opacities and thickening of the interlobular septa. Additionally, bronchiectasis and reticulations were seen. All findings are compatible with a chronic interstitial lung disease seen in alveolar proteinosis. (iv) Lung biopsy sample with protein rich exudates (line), interstitial broadening with fibroblast proliferations and scattered lymphocytes (spacing line), and hyperplasia of type II pneumocytes (arrowhead). Patient age 50 years old. Hematoxylin and eosin stain, magnification ×10; Olympus BX-51 microscope and Zeiss AxioCamMR3 camera. (B) Inherited AML/MDS family. (i) Inherited AML/MDS pedigree. Circle, female; square, male; filled red circles, history of AML/MDS; pale blue box, history of thrombocytopenia; blue striped box, normal blood counts (no further workup); unfilled boxes, individuals reported to have normal blood counts. II-1 and III-2 had ptosis. “( )”, adopted-out of the family; “+”, GATA2 wild-type; “-”, p.Thr117= GATA2. (ii) Dysplastic megakaryocytes characterized as mononuclear and with separated nuclear lobes. Pre-PBSCT marrow aspirate from II-1. Wright-Giemsa stain, magnification ×40; Leica DM 2500 microscope and MC170 HD camera. COP, cryptic organizing pneumonia; DVT, deep vein thrombosis; HCV, hepatitis C virus; HPV, human papillomavirus; VIN, vaginal intraepithelial neoplasia.
A 22-year-old woman with no antecedent cytopenias presented with AML with monosomy 7 and was treated with chemotherapy and a HLA-matched unrelated donor peripheral blood hematopoietic stem cell transplant (PBSCT, III-1; Figure 1B). She had a history of respiratory syncytial virus infection requiring hospitalization as a child but no history of recurrent infections. Her family history was notable for MDS diagnosed in her mother (II-1) at 40 years old and for MDS in a maternal aunt (II-2) at 18 years old who died of infectious complications 7 years after a HLA-matched sibling PBSCT from her affected sister. Her half-brother (III-3) has a hypocellular marrow and mild thrombocytopenia. Review of the pretransplant marrow smears of II-1 showed dysplastic megakaryocytes characterized as mononuclear and with separated nuclear lobes (Figure 1B) as described previously in GATA2 haploinsufficiency.18 Two family members (II-1, III-2) had history of ptosis corrected surgically, an extrahematopoietic phenotype reported in GATA2 deficiency19 (additional clinical history in supplemental Data).
Given their clinical histories and workups, GATA2 haploinsufficiency was suspected in both the singleton presenting with immunodeficiency and the family with multiple members diagnosed with AML or MDS. Sanger sequencing of the coding exons of GATA2 including exon-intron boundaries of genomic DNA isolated from the peripheral blood of the immunodeficiency patient revealed a novel, synonymous mutation in GATA2 exon 3 (GATA2 chr3:128,205,090G>C hg19; c.351C>G, NM_032638.4; p.Thr117=; supplemental Figure 1A).· A comprehensive screen for mutations in other known inherited bone marrow failure and inherited AML/MDS predisposition genes using a previously published11 and subsequently updated targeted capture and next generation sequencing panel was negative apart from this mutation. The same mutation was present in all family members tested from the inherited AML/MDS family (II-1, III-1, III-2, III-3; supplemental Data). Our sequencing data suggest that the GATA2 p.Thr117= mutation occurred as independent events in the 2 families reported here (supplemental Data). This mutation was absent in >1500 patients clinically suspected of having an inherited marrow failure syndrome or inherited leukemia/MDS predisposition syndrome sequenced by targeted capture methods11 and an additional cohort of 26 adult patients with immunodeficiency in whom there was a high clinical suspicion of GATA2 deficiency syndrome by Sanger sequencing of the GATA2 gene (data not shown). This mutation was reported in a study of 723 children and adolescents with primary MDS and 28 individuals clinically suspected of having GATA2 haploinsufficinency.20
The cytosine at position 351 of GATA2 complementary DNA (cDNA) is evolutionarily highly conserved among 23 mammalian species (www.ensembl.org) with a genomic evolutionary rate profiling (GERP) score of 3.27 and is absent in control databases (dbSNP and gnomAD). In silico analyses of the mutated exon 3 and its adjacent introns using various splicing algorithms (supplemental Table 1) predicted the existence of an exonic splicing enhancer and activation of a cryptic splice site. To investigate this, we performed reverse transcription-polymerase chain reaction (RT-PCR) on cultured skin fibroblasts from the immunodeficiency patient and an AML sample from the proband in the AML/MDS family (patient III-1) (supplemental Table 2; supplemental Figure 1B). Analyses of the RT-PCR products in both samples showed an additional ∼100 bp smaller PCR product in the patient samples, which was absent in controls, consistent with aberrant splicing (Figure 2A-B). To characterize the smaller PCR product, we cloned the RT-PCR products into Escherichia coli and sequenced the resultant clones. In both the fibroblast and AML samples, we generated clones expressing wild-type GATA2 transcript, GATA2 transcript containing the c.351C>G mutation, and an aberrantly spliced GATA2 transcript with a 136-bp internal deletion (Figure 2A-B; supplemental Figure 1C). To further confirm the presence of the aberrantly spliced messenger RNA (mRNA) transcript, we designed RT-PCR primers to specifically amplify the aberrantly spliced GATA2 transcript. As expected, a PCR product was only detectable in the patient sample (Figure 2C). The aberrantly spliced transcript is generated by the activation of a cryptic splice donor site at cDNA position 351, removing a 136-bp sequence between it and a cryptic splice acceptor site 3′ downstream in exon 3 (cDNA position 487). The resulting frameshift introduces a premature stop codon after 517-bp at amino acid residue 172 of 480 (Figure 2D).
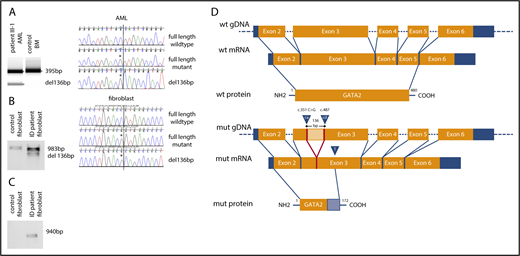
The synonymous mutation c.351C>G (p.Thr117=) in GATA2 leads to GATA2 haploinsufficiency. (A) Agarose gel electrophoresis of RT-PCR product spanning exons 3 to 4 from an AML sample from patient III-1 and a control bone marrow (BM) sample and chromatograms of PCR products. Asterisk indicates mutation/aberrant splice site. (B) Agarose gel electrophoresis of RT-PCR product spanning GATA2 exons 2 to 5 from cultured skin fibroblasts from the immunodeficiency patient (ID) and control cultured skin fibroblasts, as well as chromatograms of PCR products. (C) Agarose gel electrophoresis of RT-PCR product specific for aberrant splice product from cultured skin fibroblasts from the ID patient and control cultured skin fibroblasts. (D) Schematic overview of aberrant splicing product and putative truncated GATA2 protein generated by GATA2 c.351C>G. Blue arrow labeled with “ca” indicates cryptic splice acceptor activated by the mutation; blue arrow labeled with “cd” indicates cryptic splice donor; blue arrow labeled with “T” indicates premature stop at cDNA position 571; light orange box indicates deleted 136-bp mRNA; light blue box indicates stretch of 55 altered amino acids.
The synonymous mutation c.351C>G (p.Thr117=) in GATA2 leads to GATA2 haploinsufficiency. (A) Agarose gel electrophoresis of RT-PCR product spanning exons 3 to 4 from an AML sample from patient III-1 and a control bone marrow (BM) sample and chromatograms of PCR products. Asterisk indicates mutation/aberrant splice site. (B) Agarose gel electrophoresis of RT-PCR product spanning GATA2 exons 2 to 5 from cultured skin fibroblasts from the immunodeficiency patient (ID) and control cultured skin fibroblasts, as well as chromatograms of PCR products. (C) Agarose gel electrophoresis of RT-PCR product specific for aberrant splice product from cultured skin fibroblasts from the ID patient and control cultured skin fibroblasts. (D) Schematic overview of aberrant splicing product and putative truncated GATA2 protein generated by GATA2 c.351C>G. Blue arrow labeled with “ca” indicates cryptic splice acceptor activated by the mutation; blue arrow labeled with “cd” indicates cryptic splice donor; blue arrow labeled with “T” indicates premature stop at cDNA position 571; light orange box indicates deleted 136-bp mRNA; light blue box indicates stretch of 55 altered amino acids.
GATA2 deficiency syndrome is an increasingly diagnosed inherited cancer predisposition syndrome with multiple clinical phenotypes. In a recent study of children and young adults with primary MDS, 7% (28/426) carried germ line GATA2 mutations, with an increased incidence among those with monosomy 7 (37%).9 Genetic testing in patients with idiopathic bone marrow failure employing a targeted capture assay11 or by whole exome sequencing21 found heterozygous germ line mutations in GATA2 in ∼7% and 3% of patients, respectively. Three major classes of germ line mutations are recognized in ∼350 reported cases of GATA2 deficiency: (1) protein truncating (stop-gain, frameshift, splice-site) or large deletions, (2) missense within or adjacent to the second zinc finger domain, and (3) noncoding substitutions in intron 4 (NM_032638.4). In the few patients with canonical splice site mutations, the 5′ (c.1018-1 G>T) or 3′ exon-intron boundary of exon 5 (c.1143+5G>A) is affected.6,22 Here, we describe a novel synonymous exonic mutation not adjacent to an exon-intron junction affecting splicing of GATA2 resulting in haploinsufficiency, the major mechanism of disease. This synonymous mutation decreases GATA2 dosage by aberrant mRNA splicing of the mutated allele introducing an early stop codon. We hypothesize that this increases the susceptibility of the aberrantly spliced mRNA to nonsense mediated decay. If a protein were translated, it would lack the DNA binding domains of GATA2. Our finding establishes that synonymous substitutions in GATA2, believed thus far to be silent, warrant further evaluation by splice prediction tools and follow-up RNA analyses.
Presented in abstract form at the 59th annual meeting of the American Society of Hematology, Atlanta, GA, 10 December 2017.
The online version of this article contains a data supplement.
Acknowledgments
The authors thank Kerstina Melkaoui, Ruth Dräger, and Bettina Wehrle for technical assistance.
This work was supported by grants from the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health (R24 DK099808) (M.-C.K.) and Federal Ministry of Education and Research (BMBF 01EO1303) (C.W. and U.S.). M.-C.K. is an American Cancer Society professor.
Authorship
Contribution: C.W., K.G., S.C., D.B., M.S., S.G., S.K., M.-C.K., C.C.P., T.W., D.W., and U.S. performed experiments and interpreted data; S.F.N.B., B.C.F., S.K., and M.-B.P. contributed patient samples and clinical characterizations; and C.W., S.K., T.W., D.W., and U.S. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Ulrich Salzer, Center for Chronic Immunodeficiency (CCI) and Department of Rheumatology and Clinical Immunology, Medical Center–University of Freiburg, Faculty of Medicine, University of Freiburg, Hugstetterstr 55, 79106 Freiburg, Germany; e-mail: ulrich.salzer@uniklinik-freiburg.de.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal