

Key Points
T cells expressing a CAR consisting of scFv #213 targeting WT1 peptide/HLA-A*2402 complex killed HLA-A*2402+ WT1+ tumor cell lines.
The therapeutic efficacy of #213 scFv CAR-T cells was shown to be enhanced by DC vaccine in a murine xenograft model.

Abstract
The recent success of chimeric antigen receptor (CAR)-T cell therapy for treatment of hematologic malignancies supports further development of treatments for both liquid and solid tumors. However, expansion of CAR-T cell therapy is limited by the availability of surface antigens specific for the tumor while sparing normal cells. There is a rich diversity of tumor antigens from intracellularly expressed proteins that current and conventional CAR-T cells are unable to target. Furthermore, adoptively transferred T cells often suffer from exhaustion and insufficient expansion, in part, because of the immunosuppressive mechanisms operating in tumor-bearing hosts. Therefore, it is necessary to develop means to further activate and expand those CAR-T cells in vivo. The Wilms tumor 1 (WT1) is an intracellular oncogenic transcription factor that is an attractive target for cancer immunotherapy because of its overexpression in a wide range of leukemias and solid tumors, and a low level of expression in normal adult tissues. In the present study, we developed CAR-T cells consisting of a single chain variable fragment (scFv) specific to the WT1235-243/HLA-A*2402 complex. The therapeutic efficacy of our CAR-T cells was demonstrated in a xenograft model, which was further enhanced by vaccination with dendritic cells (DCs) loaded with the corresponding antigen. This enhanced efficacy was mediated, at least partly, by the expansion and activation of CAR-T cells. CAR-T cells shown in the present study not only demonstrate the potential to expand the range of targets available to CAR-T cells, but also provide a proof of concept that efficacy of CAR-T cells targeting peptide/major histocompatibility complex can be boosted by vaccination.
Introduction
Adoptive transfer of T cells genetically modified to express an artificial receptor consisting of the variable fragment of an antibody specific for a cell surface molecule linked to a T-cell signaling molecule, termed chimeric antigen receptor (CAR), is emerging as a promising approach for cancer immunotherapy. A CAR consists of a single chain variable fragment (scFv) as an ectodomain, a short hinge, a transmembrane domain, and an endodomain with signaling domains derived from CD3ζ and costimulatory molecules. Recent clinical trials of adoptive therapy with CAR-T cells targeting CD19 have shown an impressive efficacy in patients with hematologic malignancies,1-5 which suggests that this approach may be extended to treat common epithelial cancers. However, wider application of CAR-T cell therapy is limited by the availability of cell surface tumor associated antigens (TAAs) specific for the tumor while sparing normal cells. There is a rich diversity of tumor antigens from intracellularly expressed proteins that act as disease drivers, such as oncogene products, or are expressed in tumor cells exclusively or in very specific tissues, such as neoantigens or the CT antigens, respectively.6 Furthermore, recent clinical trials explored the potential of posttransfer vaccination to enhance clinical efficacy of adoptively transferred T cells expressing T-cell receptor (TCR) specific for an intracellular tumor antigen in the context of major histocompatibility complex (MHC) class I.7-13 Recent studies also have demonstrated that CAR-T cells specific for a cell surface TAA also expressing endogenous TCR specific for a strong immunogen (dual specific T cells) displayed robust expansion and antitumor activity against tumor expressing the corresponding TAA upon vaccination with the immunogen.14-16 However, the exploitation of dual specific T cells involves intricate processes, and posttransfer vaccination is hardly applicable for current CAR-T cells because of their inability to recognize peptide antigen in the context of MHC. In those respects, a TCR-based targeting approach is more suitable in that it targets peptides in association with the MHC derived from intracellular proteins on tumor cells and cross-presented by antigen presenting cells; thus, adoptive cell therapy based on TCR-engineered T cells can be enhanced by vaccination. However, further development of this approach is hampered because of difficulty in TCR acquisition and the inherent low affinity of isolated TCR for the peptide/MHC complex.17-19 Attempts to artificially increase the affinity of isolated TCR in vitro sometimes resulted in cross-reactivity to endogenous self-peptide/MHC.20,21 To circumvent these constraints, CARs consisting of an scFv that recognizes peptide/MHC have been developed.22 Using a phage-display library, a monoclonal antibody against peptide-MHC complexes could be isolated entirely in vitro with efficiency surpassing that of isolation of TCR.23,24
The ideal target antigen for adoptive cell therapy should be expressed in a cancer-specific fashion to avoid damaging healthy tissues and be intrinsic to cancer cells so that downregulation of the antigen by the tumor cannot occur. The zinc finger transcription factor, Wilms’ tumor-1 (WT1) protein, is an attractive target because it has an important role in cell growth and differentiation; is overexpressed in leukemias, lymphomas, and solid tumors; and has limited expression in normal tissues.25,26 Although WT1 was originally identified as a tumor suppressor,27,28 its oncogenic role in leukemogenesis and tumorigenesis became evident over time.29 Overexpression of WT1 has been demonstrated in the CD34+ stem cells of patients with chronic and acute myeloid leukemia,30,31 making it an attractive therapeutic target for eradication of these diseases. The amino acid residues 235-243 (CMTWNQMNL) of the WT1 protein were previously validated as an epitope expressed in human leukemia cells and epithelial cancer cells in association with HLA-A*2402,32,33 a predominant type of HLA-A allele in Asian populations (http://www.allelefrequencies.net),34 which provides a rationale for targeting this epitope with a CAR. However, this 9-mer peptide lacks a second position anchor motif, which makes it unstable as a peptide/HLA complex. Given that anchor residues are usually buried in the MHC cleft and not exposed to the surface of a peptide/MHC complex, vaccination with a modified WT1 peptide, WT1236Y, in which an amino acid substitution, M→Y at position 2, was introduced to increase MHC binding, has been shown to successfully induce T cells to recognize the natural WT1 peptide on WT1+/HLA-A*2402+ tumors.35 Therefore, we employed a WT1236Y loaded on HLA-A*2402 for the isolation of an scFv specific to WT1235-243/HLA-A*2402. Here, we describe the generation and therapeutic efficacy of T cells modulated to express a CAR that consists of an scFv specific to the WT1235-243/HLA-A*2402 complex.
Materials and methods
All materials and methods are described in the supplemental Materials, available on the Blood Web site.
Results
Antigen-specific cytotoxic activity of #213 scFv CAR-transduced T cells against peptide-pulsed HLA-A*2402-transduced T2 cell line
Isolated scFv reactive to WT1236Y/HLA-A*2402 but not to HLA-A*2402 or an irrelevant peptide/HLA-A*2402 from clone #213 (supplemental Figure 1A) was inserted into retrovirus vector pMS3 linking the intracellular signaling domains of CD3ζ and GITR (Figure 1A) and transduced into anti-CD3-stimulated human peripheral blood mononuclear cells (PBMCs). Both CD4 and CD8 T cells expressed CAR, as judged by WT1236Y/HLA-A*2402 tetramer binding (Figure 1B). Afterward, #213 scFv CAR-T cells were examined for their antigen-specific activation using HLA-A*2402 tetramer loaded with relevant or irrelevant peptides. As shown in Figure 1C, #213 scFv CAR-T cells produced interferon-γ (IFN-γ) upon stimulation by immobilized HLA-A*2402 tetramer loaded with WT1235-243 or WT1236Y peptide in a dose-dependent manner, but to a lesser extent in WT1235-243. On the other hand, #213 scFv CAR-T cells did not produce IFN-γ upon incubation with HLA-A*2402 tetramer loaded with MAGE-A4143-151 peptide. This antigen-specific activation of #213 scFv CAR-T cells was further confirmed by activity against T2A24 cells pulsed with WT1235-243 or WT1236Y, but again to a lesser extent in WT1235-243, and not against cells nonpulsed or pulsed with irrelevant peptide (Figure 1D). This WT1/HLA-A*2402-specific lysis by CAR-T cells was accompanied by an increase in IFN-γ production upon cultivation with T2A24 cells pulsed with WT1236Y or WT1235-243, but not with irrelevant peptide, as assessed by intracellular cytokine staining (Figure 1E).
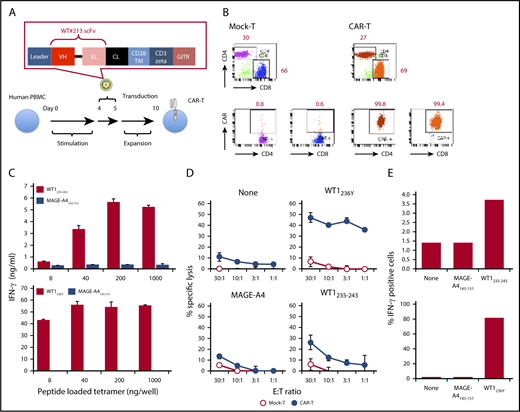
Design and characterization of a WT1235-243/HLA-A*2402-specific CAR. (A) Schematic representation of a retroviral vector encoding WT1235-243/HLA-A*2402-specific CAR. (B) Expression of CAR on human T cells. Human PBMCs stimulated with plate coated anti-CD3 were transduced with the retroviral vector as depicted in panel A. Four days after the transduction, cells were stained with phycoerythrin-labeled HLA-A*2402 tetramers presenting WT1236Y along with APC anti-CD4 and APC/Cy7 anti-CD8. Mock-transduced T cells served as background staining. (C) Production of IFN-γ by #213 scFv CAR-T cells stimulated with immobilized HLA-A*2402 tetramers loaded with WT1235-243 or WT1236Y. Culture supernatants were harvested at 24 hours and subjected to IFN-γ enzyme-linked immunosorbent assay in triplicate. (D) Cytotoxic activity of #213 scFv CAR-T cells against T2A24 cells pulsed with the indicated peptides at 10 μM was assayed by 6-hour 51Cr-release assays in triplicate. (E) IFN-γ production of #213 scFv CAR-T cells stimulated with T2A24 cells pulsed with the indicated peptides. % IFN-γ–positive cells within CD8+ cells of #213 scFv CAR-T cells were assayed by intracellular cytokine staining after a 6-hour culture in single experimental samples. Error bars represent standard deviation (SD) of the mean. A representative result of 3 independent experiments is shown.
Design and characterization of a WT1235-243/HLA-A*2402-specific CAR. (A) Schematic representation of a retroviral vector encoding WT1235-243/HLA-A*2402-specific CAR. (B) Expression of CAR on human T cells. Human PBMCs stimulated with plate coated anti-CD3 were transduced with the retroviral vector as depicted in panel A. Four days after the transduction, cells were stained with phycoerythrin-labeled HLA-A*2402 tetramers presenting WT1236Y along with APC anti-CD4 and APC/Cy7 anti-CD8. Mock-transduced T cells served as background staining. (C) Production of IFN-γ by #213 scFv CAR-T cells stimulated with immobilized HLA-A*2402 tetramers loaded with WT1235-243 or WT1236Y. Culture supernatants were harvested at 24 hours and subjected to IFN-γ enzyme-linked immunosorbent assay in triplicate. (D) Cytotoxic activity of #213 scFv CAR-T cells against T2A24 cells pulsed with the indicated peptides at 10 μM was assayed by 6-hour 51Cr-release assays in triplicate. (E) IFN-γ production of #213 scFv CAR-T cells stimulated with T2A24 cells pulsed with the indicated peptides. % IFN-γ–positive cells within CD8+ cells of #213 scFv CAR-T cells were assayed by intracellular cytokine staining after a 6-hour culture in single experimental samples. Error bars represent standard deviation (SD) of the mean. A representative result of 3 independent experiments is shown.
#213 scFv CAR-transduced T cells lyse endogenously WT1-expressing tumor cell lines in an HLA-A*2402–dependent manner
In the next series of experiments, we determined whether #213 scFv CAR-T cells could react to endogenously derived WT1 peptide in association with HLA-A*2402. First, a panel of tumor cell lines was used as targets for an intracellular cytokine staining assay. As shown in Figure 2A, #213 scFv CAR-T cells produced IFN-γ upon interaction with WT1+HLA-A*2402+ tumor cell lines, such as HMMME, MESO-4, and K562 transfected with HLA-A*2402 (K562-A24), but not with tumor cell lines lacking either or both of WT1+ and HLA-A*2402, such as TE4, MESO-1, K562, HeLa, and LK-2. Although IFN-γ–positive cell number within #213 scFv CAR-T cells was further increased upon incubation with WT1+HLA-A*2402+ tumor cell lines treated with IFN-γ to increase HLA expression and immunoproteasome activation, the pattern of #213 scFv CAR-T cell reactively toward those cell lines remained largely the same (data not shown). Mock-transduced T cells did not demonstrate any reactivity against all of these tumor cell lines (data not shown). Similarly, #213 scFv CAR-T cells also demonstrated cytotoxic activity toward WT1+HLA-A*2402+ tumor cell lines, but not toward those lacking of WT1+ or HLA-A*2402 expression (Figure 2B). To further confirm the epitope specificity of #213 CAR-T cells against WT1+ tumor cells, a standard 51Cr release assay using MESO-4 cell line as a target at an effector-to-target ratio of 2:1 was conducted in the presence of 40-fold excess unlabeled T2A24 pulsed with either WT1236Y, WT1235-243, or MAGE-A4143-151. As shown in Figure 2C, the lysis of MESO-4 cells by #213 scFv CAR-T cells was inhibited by T2A24 cells loaded with WT1236Y, but not with WT1235-243 or MAGE-A4143-151.
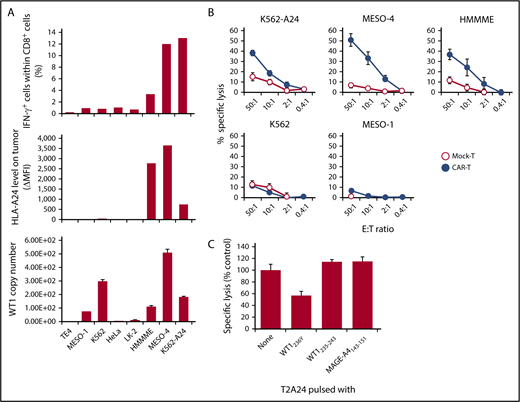
#213 scFv CAR-T cells react to tumor cell lines endogenously expressing WT1 in an HLA-A*2402–restricted manner. (A) IFN-γ production of CD8+ cells of #213 scFv CAR-T cells in response to the indicated tumor cell lines was assayed by intracellular cytokine staining after a 6-hour culture (top) along with HLA-A*2402 (middle) and WT1 mRNA expression of those tumor cell lines (bottom). (B) Cytotoxic activity of #213 scFv CAR-T cells against the indicated tumor cell lines was assessed by 6-hour 51Cr-release assays in triplicate. (C) Confirmation of epitope specificity of #213 scFv CAR-T cells against peptide derived from endogenously expressing WT1. A standard 51Cr release assay using purified CD8+CAR+ T cells as effectors and a MESO-4 cell line as a target at an effector-to-target ratio of 2:1 was conducted in the presence of 40-fold excess of unlabeled T2A24 pulsed with either WT1236Y or MAGE-A4143-151. Error bars represent SD of the mean. A representative result of 3 independent experiments is shown.
#213 scFv CAR-T cells react to tumor cell lines endogenously expressing WT1 in an HLA-A*2402–restricted manner. (A) IFN-γ production of CD8+ cells of #213 scFv CAR-T cells in response to the indicated tumor cell lines was assayed by intracellular cytokine staining after a 6-hour culture (top) along with HLA-A*2402 (middle) and WT1 mRNA expression of those tumor cell lines (bottom). (B) Cytotoxic activity of #213 scFv CAR-T cells against the indicated tumor cell lines was assessed by 6-hour 51Cr-release assays in triplicate. (C) Confirmation of epitope specificity of #213 scFv CAR-T cells against peptide derived from endogenously expressing WT1. A standard 51Cr release assay using purified CD8+CAR+ T cells as effectors and a MESO-4 cell line as a target at an effector-to-target ratio of 2:1 was conducted in the presence of 40-fold excess of unlabeled T2A24 pulsed with either WT1236Y or MAGE-A4143-151. Error bars represent SD of the mean. A representative result of 3 independent experiments is shown.
Peptide motif recognized by #213 scFv CAR
To evaluate the potential risks of cross-reactivity to self-peptides, we first sought to determine the critical amino acid residues of WT1235-243 peptide for binding of #213 CAR to the HLA-A*2402/WT1 peptide complex. An alanine substitution assay using a WT1236Y peptide sequence substituted with alanine at positions 1 to 9 revealed that the second, third, fourth, and fifth amino acid residues of the WT1235-243 peptide were critical for tighter interaction with #213 CAR-T cells (Figure 3A). We also confirmed that the anchor amino acids required for WT1 to bind HLA-A*2402 were the second and ninth amino acids by an HLA-stabilizing assay (Figure 3B). Based on these results, an in silico search was conducted using the BLAST database program, blastp (http://blast.ncbi.nlm.nih.gov/Blast.cgi) to identify protein sequences that contain xYTWNxxxL and xMTWNxxxL motifs. We included peptide motifs with F or W at the second residue; I, M, or W at the ninth residue; and 10-mer peptides, which resulted in predictably high binding affinity to HLA-A*2402 compared with that of WT1235-243 (supplemental Table 1). Furthermore, peptide motifs with amino acid residues at the third, fourth, and fifth positions substituted by those with similar physicochemical properties were tested to activate #213 CAR-T cells (supplemental Figure 4). Based on this, we performed blast search using motifs xYSWNxxxx, xYNWNxxxx, xYTFNxxxx, xYTWHxxxx, xWTWNxxxx, xWSWNxxxx, xWNWNxxxx, xWTFNxxxx, and xWTWHxxxx, and obtained 38 peptides from human proteome. Three (including 1 coded by pseudo gene) of 38 peptides loaded on T2A24 induced IFN-γ production by #213 CAR-T cells (Figure 3C). Additionally, we further explored the possible cross-reactivity against allo-HLA molecules using a panel of lymphoblastoid cell lines (LCLs) expressing different HLA molecules. As shown in Figure 3D, none of LCLs expressing different allo-HLA molecules did not stimulate #213 scFv CAR-T cells to produce IFN-γ.
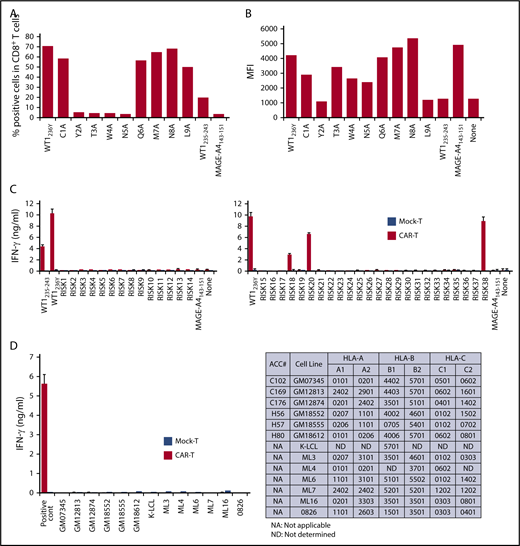
Potential cross-reactivity of #213 scFv CAR-T cells to homologous peptides on HLA-A*2402. (A) Alanine substitution analysis identified WT1235 peptide residues important for recognition by #213 scFv CAR. The WT1236Y peptide sequence was substituted with alanine from 1 through 9. T2A24 was pulsed with the indicated peptides at 10 μM and cocultured with #213 scFv CAR-T cells for 6 hours. IFN-γ–positive cells gated on CD8+ cells in CAR-T cells were assayed by intracellular cytokine staining. (B) The peptides in panel A were subjected to an HLA stabilization assay to determine the critical amino acids required for HLA-A*2402 binding. T2A24 cells were used as peptide-loading cells and their HLA-A*2402 expression was determined by fluorescence-activated cell sorter (FACS). (C) Validation of potential risk peptides derived from the human proteome. T2A24 cells were pulsed with the indicated peptides (sequences provided in supplemental Table 1) at 10 μM and cocultured with #213 scFv CAR-T cells (black column) or mock-transduced T cells (white column) for 24 hours. (D) Validation of potential cross-reactivity to allo-HLAs. LCLs expressing different HLAs were cocultured with #213 scFv CAR-T cells (black column) or mock-transduced T cells (white column) for 24 hours. T2A24 cells pulsed with 10 μM WT1236Y served as a positive control. Culture supernatants from these cultures were subjected to IFN-γ enzyme-linked immunosorbent assay in triplicate. Error bars represent SD of the mean. A representative result of 3 independent experiments is shown.
Potential cross-reactivity of #213 scFv CAR-T cells to homologous peptides on HLA-A*2402. (A) Alanine substitution analysis identified WT1235 peptide residues important for recognition by #213 scFv CAR. The WT1236Y peptide sequence was substituted with alanine from 1 through 9. T2A24 was pulsed with the indicated peptides at 10 μM and cocultured with #213 scFv CAR-T cells for 6 hours. IFN-γ–positive cells gated on CD8+ cells in CAR-T cells were assayed by intracellular cytokine staining. (B) The peptides in panel A were subjected to an HLA stabilization assay to determine the critical amino acids required for HLA-A*2402 binding. T2A24 cells were used as peptide-loading cells and their HLA-A*2402 expression was determined by fluorescence-activated cell sorter (FACS). (C) Validation of potential risk peptides derived from the human proteome. T2A24 cells were pulsed with the indicated peptides (sequences provided in supplemental Table 1) at 10 μM and cocultured with #213 scFv CAR-T cells (black column) or mock-transduced T cells (white column) for 24 hours. (D) Validation of potential cross-reactivity to allo-HLAs. LCLs expressing different HLAs were cocultured with #213 scFv CAR-T cells (black column) or mock-transduced T cells (white column) for 24 hours. T2A24 cells pulsed with 10 μM WT1236Y served as a positive control. Culture supernatants from these cultures were subjected to IFN-γ enzyme-linked immunosorbent assay in triplicate. Error bars represent SD of the mean. A representative result of 3 independent experiments is shown.
In vivo therapeutic efficacy of #213 scFv against WT1+/HLA-A*2402+ tumors
Next, we proceeded to evaluate the therapeutic potential of #213 scFv CAR-T cells in vivo in a xenograft model using immunodeficient NOG mice transplanted with K562 (WT1+/HLA-A*2402−) or K562 transfected with HLA-A*2402 (K562-A24, WT1+/HLA-A*2402+). As shown in Figure 4, adoptive transfer of #213 scFv CAR-T cells into NOG mice that were injected subcutaneously with K562 or K562-A24 on the same day suppressed growth of K562-A24, but not K562, which indicates that #213 scFv CAR-T cells exerted an antitumor effect in vivo in an antigen-specific manner.
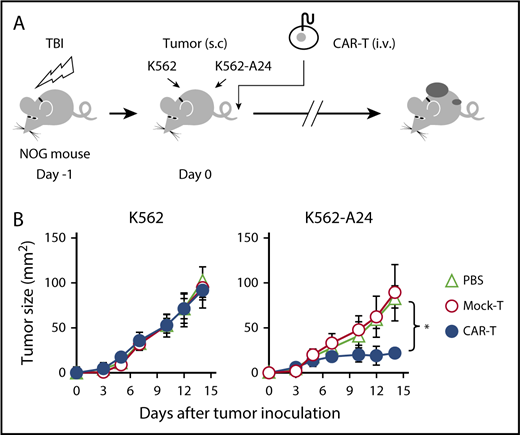
Adoptive transfer of WT1-specific CAR-T cells suppressed tumor growth in an antigen-specific manner. (A) Schematic representation for the adoptive transfer experiment using NOG mice. (B) Tumor growth curves of K562 and K562-A24 in NOG mice (n = 4) transferred with CAR-T cells or mock transduced T cells. NOG mice were inoculated s.c. with K562 and K562-A24 (5 × 106 cells) followed by IV injection with #213 CAR-T cells or mock transduced T cells (1 × 107 cells). Tumor volumes were measured by a caliper using the formula (length × width) at the indicated time points (n = 5). Error bars represent SD of the mean. *P < .05. A representative result from 2 independent experiments is shown. s.c., subcutaneous; TBI, total body irradiation.
Adoptive transfer of WT1-specific CAR-T cells suppressed tumor growth in an antigen-specific manner. (A) Schematic representation for the adoptive transfer experiment using NOG mice. (B) Tumor growth curves of K562 and K562-A24 in NOG mice (n = 4) transferred with CAR-T cells or mock transduced T cells. NOG mice were inoculated s.c. with K562 and K562-A24 (5 × 106 cells) followed by IV injection with #213 CAR-T cells or mock transduced T cells (1 × 107 cells). Tumor volumes were measured by a caliper using the formula (length × width) at the indicated time points (n = 5). Error bars represent SD of the mean. *P < .05. A representative result from 2 independent experiments is shown. s.c., subcutaneous; TBI, total body irradiation.
Activation of #213 scFv CAR-T cells by APCs loaded with WT1236Y in a HLA-A*2402-restricted manner
In the final series of experiments, we sought to determine whether the antitumor function of #213 scFv CAR-T cells in vivo could be enhanced by boosting immunization. To this end, we first tested whether a corresponding peptide WT1236Y on APCs could induce activation of #213 scFv CAR-T cells in an HLA-A*2402–restricted manner. In this case, PBMCs depleted of CD3+ cells from HLA-A*2402+ or HLA-A*2402− donors pulsed with WT1236Y or synthetic WT1236Y long peptide (LP) were used as stimulator. We confirmed that #213 scFv CAR-T cells prepared from 3 different donors produced IFN-γ upon interaction with APC loaded with the corresponding antigen in an HLA-A*2402–restricted manner (supplemental Figure 5).
Enhancement of the therapeutic efficacy of #213 scFv CAR-T cells by vaccination with DCs loaded with WT1236Y
Immunodeficient mice reconstituted with PBMCs (humanized mice) are considered the most amenable for studying human TCRs upon vaccination. However, immune-deficient mice are highly susceptible to TLR agonists co-opted by an adjuvant, which causes rapid death due to a cytokine storm.36 To circumvent these constraints, we used in vitro generated mature human dendritic cells (DCs) pulsed with an antigenic peptide for vaccination. We confirmed that our DC preparation (supplemental Figure 6) loaded with WT1236Y, but not with MAGE-A4143-151, efficiently induced proliferation of #213 scFv CAR-T cells in a carboxyfluorescein diacetate succinimidyl ester (CFSE)-dilution assay (Figure 5). Next, NOG mice bearing 3-day-old K562-A24 tumors were transferred with #213 scFv CAR-T cells or CEA-specific CAR-T cells (F11-39 scFv CAR-T cells)37 serving as an additional control, followed immediately by intravenous vaccination with 1 × 105 mature human DCs pulsed with WT1236Y. In this therapeutic setting, the growth of K562-A24 was not suppressed by the transfer of #213 scFv CAR-T cells alone, but concurrent intravenous vaccination with DCs pulsed with WT1236Y, but not with MAGE-A4143-151, resulted in significant suppression of K562-A24 growth (Figure 6). In addition, the therapy was ineffective when transferred T cells express a CEA-specific CAR.
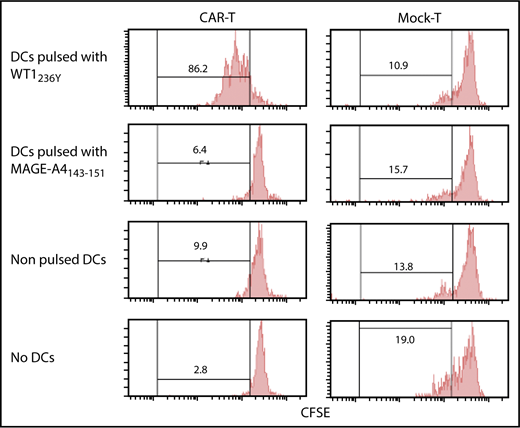
Proliferation of #213 scFv CAR-T cells in response to WT1236Y directly presented by DCs. CFSE-labeled #213 scFv CAR-T cells were cocultured with HLA-A*2402+ DCs with WT1236Y peptide (WT1236Y SP) and subjected to CFSE-dilution assay on day 3. DCs pulsed with 10 μM MAGE-A4 peptides served as a negative control. A representative result from 3 independent experiments is shown.
Proliferation of #213 scFv CAR-T cells in response to WT1236Y directly presented by DCs. CFSE-labeled #213 scFv CAR-T cells were cocultured with HLA-A*2402+ DCs with WT1236Y peptide (WT1236Y SP) and subjected to CFSE-dilution assay on day 3. DCs pulsed with 10 μM MAGE-A4 peptides served as a negative control. A representative result from 3 independent experiments is shown.
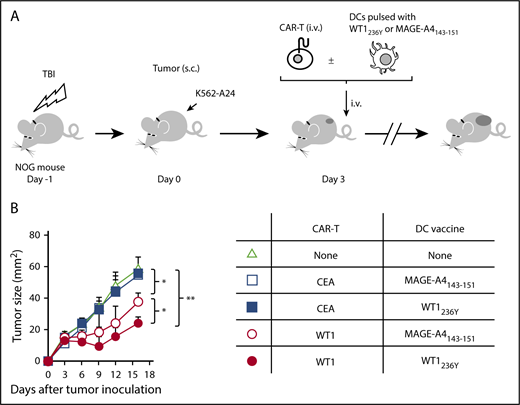
Vaccination with DCs pulsed with WT1236Y enhanced WT1-specific CAR-T cells to suppress growth of WT1-positive tumors. (A) Schematic representation for the adoptive transfer experiment using NOG mice. (B) Tumor growth curves of K562-A24 in NOG mice (n = 4) transferred with CAR-T cells together with or without DCs pulsed with a relevant or irrelevant peptide. NOG mice bearing 3-day-old K562-A24 tumors were transferred with #213 scFv CAR-T cells or CEA-specific (F11-39 scFv) CAR-T cells (1 × 107 cells) together with or without DCs (1 × 105 cells) pulsed with WT1236Y or MAGE-A4143-151. Tumor volumes were measured by a caliper using the formula (length × width) at the indicated time points. Error bars represent SD of the mean. *P < .05; **P < .01. A representative result from 2 independent experiments is shown.
Vaccination with DCs pulsed with WT1236Y enhanced WT1-specific CAR-T cells to suppress growth of WT1-positive tumors. (A) Schematic representation for the adoptive transfer experiment using NOG mice. (B) Tumor growth curves of K562-A24 in NOG mice (n = 4) transferred with CAR-T cells together with or without DCs pulsed with a relevant or irrelevant peptide. NOG mice bearing 3-day-old K562-A24 tumors were transferred with #213 scFv CAR-T cells or CEA-specific (F11-39 scFv) CAR-T cells (1 × 107 cells) together with or without DCs (1 × 105 cells) pulsed with WT1236Y or MAGE-A4143-151. Tumor volumes were measured by a caliper using the formula (length × width) at the indicated time points. Error bars represent SD of the mean. *P < .05; **P < .01. A representative result from 2 independent experiments is shown.
Enhanced therapeutic efficacy of #213 scFv CAR-T cells by DC vaccination is associated with the expansion and activation of transferred CAR-T cells
To obtain mechanistic insight into how DC vaccine enhances antitumor efficacy of CAR-T cells targeting peptide/MHC, we investigated the abundance and/or activation status of CAR-T cells in tumor tissues and peripheral blood of mice received DCs pulsed with WT1236Y or MAGE-A4143-151. As shown in Figure 7A, the frequency of #213 scFv CAR-T cells was increased in mice treated with DC/WT1236Y vaccination compared with those treated with DC/MAGE-A4143-151 vaccination, indicative of antigen-specific accumulation in the periphery. Furthermore, tumor tissues from the mice vaccinated with DC/WT1236Y but not DC/MAGE-A4143-151 were infiltrated with activated #213 scFv CAR-T cells as judged by CD8+ cells coexpressing MHC class II (Figure 7B).38,39 Because #213 scFv CAR-T and F11-39 scFv CAR-T cells are unable to be stained by tetramer or biotinylated-CEA, respectively, in tissue sections, we used CD8 as a surrogate marker for CAR-T cells. In line with these results, peripheral accumulation of CAR-T cells (Figure 8B) associated with improved antigen-specific tumor control was also observed in mesothelioma-bearing NOG mice receiving DC vaccine (Figure 8A). In another set of experiments, the number and frequency of Ki67+CD8+ cells indicative of proliferating CAR-T cells significantly increased in the tumor tissue of MESO-4 bearing mice received the DC/WT1236Y vaccination (Figure 8C; supplemental Figure 7). However, there was a tendency of increased numbers of CAR-T cells only. Collectively, these results show that a posttransfer vaccination can induce substantially stronger antitumor effects of CAR-T cells targeting intracellular tumor antigens possibly via CAR-T cell expansion and activation.
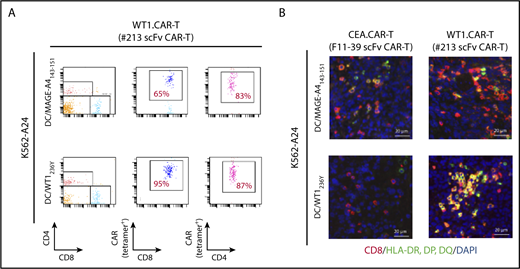
Antigen-specific DC vaccination enhanced #213 scFv CAR-T cells accumulation in periphery and activation in tumor tissues. Three-day K562-A24 bearing NOG mice were transferred with #213 scFv CAR-T cells or CEA-specific (F11-39 scFv) CAR-T cells (1 × 107 cells) followed by vaccination with DCs (1 × 105 cells) pulsed with WT1236Y or MAGE-A4143-151 as Figure 6. On day 16, PBMCs and tumor tissues were harvested and subjected to FACS (A) and immunohistochemical (B) analysis, respectively. A representative result from 2 independent experiments is shown.
Antigen-specific DC vaccination enhanced #213 scFv CAR-T cells accumulation in periphery and activation in tumor tissues. Three-day K562-A24 bearing NOG mice were transferred with #213 scFv CAR-T cells or CEA-specific (F11-39 scFv) CAR-T cells (1 × 107 cells) followed by vaccination with DCs (1 × 105 cells) pulsed with WT1236Y or MAGE-A4143-151 as Figure 6. On day 16, PBMCs and tumor tissues were harvested and subjected to FACS (A) and immunohistochemical (B) analysis, respectively. A representative result from 2 independent experiments is shown.
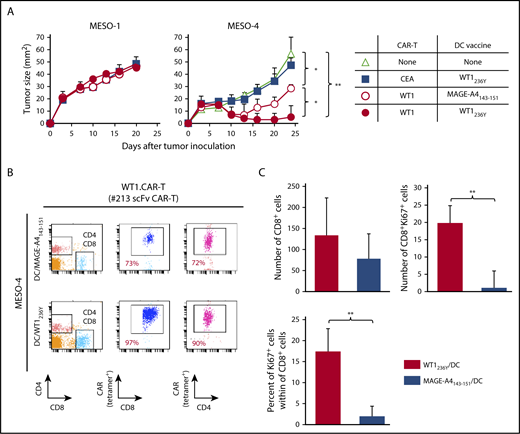
Adoptive transfer with #213 scFv CAR-T cells suppressed growth of mesothelioma cell line, which was enhanced by the DC vaccine. Three-day MESO-1– or MESO-4–bearing NOG mice (n = 4) were transferred with #213 scFv CAR-T cells or CEA-specific (F11-39 scFv) CAR-T cells (1 × 107 cells) followed by vaccination with DCs (1 × 105 cells) pulsed with WT1236Y or MAGE-A4143-151. (A) Tumor volumes were measured by a caliper using the formula (length × width) at the indicated time points. (B) On day 24, PBMCs and tumor tissues were harvested and subjected to FACS analysis. In another set of experiments, tumor tissues were harvested from MESO-4 bearing NOG mice (n = 3) treated as mentioned previously on day 18 and subjected to immunohistochemical analysis. Cells with respective markers were analyzed in 6 fields, and the mean ± SD are depicted (C). Error bars represent SD of the mean. *P < .05; **P < .01. A representative result from 2 independent experiments is shown.
Adoptive transfer with #213 scFv CAR-T cells suppressed growth of mesothelioma cell line, which was enhanced by the DC vaccine. Three-day MESO-1– or MESO-4–bearing NOG mice (n = 4) were transferred with #213 scFv CAR-T cells or CEA-specific (F11-39 scFv) CAR-T cells (1 × 107 cells) followed by vaccination with DCs (1 × 105 cells) pulsed with WT1236Y or MAGE-A4143-151. (A) Tumor volumes were measured by a caliper using the formula (length × width) at the indicated time points. (B) On day 24, PBMCs and tumor tissues were harvested and subjected to FACS analysis. In another set of experiments, tumor tissues were harvested from MESO-4 bearing NOG mice (n = 3) treated as mentioned previously on day 18 and subjected to immunohistochemical analysis. Cells with respective markers were analyzed in 6 fields, and the mean ± SD are depicted (C). Error bars represent SD of the mean. *P < .05; **P < .01. A representative result from 2 independent experiments is shown.
Discussion
The recent clinical success of adoptive cell therapy using CAR-modified T cells targeting CD19 represents a paradigm shift in cancer immunotherapy. However, the wider application of this therapeutic approach is constrained by the rarity of cell surface antigens exclusively or abundantly expressed on tumor cells. In many cases, tumor-specific antigens are intracellular proteins that cannot be targeted by current CARs.40 An emerging approach to circumvent this limitation involves CARs consisting of scFvs, in which recognition of antigen is similar to that by TCRs (ie, antigen peptide in association with MHC).23 In the present study, we developed a CAR consisting of a scFv #213 directed against an amino acid sequence derived from WT1, which is overexpressed in various types of tumors, in association with HLA-A*2402, a major type of HLA-A allele of Asian populations.
The majority of cancer peptide epitopes that have been described, including WT1235-243, do not contain optimal primary peptide anchor residues generally located toward the N and C terminus.41 Therefore, for the screening and isolation of scFv-reactive WT1235-243/HLA-A*2402 from a phage display library, we adopted an approach wherein scFvs reactive to a modified WT1 peptide, WT1236Y, loaded on HLA-A*2402 tetramer were rescreened based on their reactivity to WT1235-243/HLA-A*2402. We have successfully isolated scFv #213 that binds authentic WT1235-243/HLA-A*2402. T cells transduced with a CAR consisting of #213 scFv killed a panel of WT1/HLA-A*2402-expressing tumor cell lines. The epitope specificity of #213 scFv CAR-T cells to an authentic WT1235-243 peptide in association with HLA-A*2402 was confirmed in that T2A24 cells loaded with WT1236Y efficiently inhibited lysis of the WT1+/HLA-A*2402+ tumor cell line MESO-4 by #213 scFv CAR-T cells.
An important implication of our results is that peptide WT1235-243, with a very high predicted 50% inhibitory concentration value (7889.1 nM by IEBD) on HLA-A*2402, can activate T cells with low-affinity CAR (distribution coefficient [Kd] = 741 nM) (supplemental Figure 1B). Whereas TCR has evolved to co-opt the machinery that enables exquisitely sensitive recognition and proximal signaling efficacy, CARs represent synthetic and artificial constructs with a fixed first and second signaling ratio. T cells with a TCR affinity of 100 μM can be activated by a few peptide MHC complexes on the surface of the target cell.42-45 Within the natural TCR affinity range (Kd = 2 to 200 μM) and under low ligand (cognate peptide/MHC) density, substantial evidence indicates that enhanced TCR affinity correlates with improved T-cell responsiveness.46 However, it has also been suggested that although TCR and ligand must stay bound for a sufficiently long duration to enable biochemical responses in T cells to activate, TCR must release the ligand quickly (fast off-rate) so that rare antigenic peptide MHCs have an opportunity to engage other TCRs and thus amplify signaling cascades (ie, TCRs must have an optimal affinity range to enable serial triggering).47-49 It was indeed shown that T cells bearing TCRs with artificially increased supraphysiological affinity (Kd < 1 μM) resulted in a drastic functional decline while retaining antigen specificity toward cognate peptide/MHC complexes.50-52 With regard to CARs, there is also a relationship among the CAR affinity and antigen density that affects the effector function of CAR-T cells.53 Although there is a limited information about the minimum number of cell surface antigen molecules required by CAR-T cells to activate, CARs that target ligands present on target cells with higher densities (∼5400-30 500/cells) have a reported affinity range between 0.3 and 4 nM.54-57 In sharp contrast, it also has been shown that low-affinity CAR (Kd = 1616 nM) is more effective than high-affinity CAR (Kd = 1 nM) under the conditions of limiting antigen molecules.58 Given the epitope density of a low MHC binder (for example, WT1235-243 on a target cell is presumably extremely low), it is possible that the affinity of #213 scFv (Kd = 741 nM) could be within the appropriate range for serial triggering to occur to facilitate activation of our CAR-T cells, similar to those seen in TCRs. In support of this notion, T cells transduced with CAR consisting of another scFv #5 with higher affinity to WT1235-243/A*2402 (Kd = 34.4 nM) produced IFN-γ upon incubation with T2A24 cells pulsed with WT1236Y but not with WT1+/ A*2402+ tumor cell lines. Nevertheless, this scenario remains speculative and needs to be supported with evidence obtained using different target antigens and CARs. Recent studies have started to unravel the mechanism of action of CAR,59 but the molecular explanation to clarify why and how low MHC binders, such as WT1235-243 on HLA-A*2402, can efficiently activate CAR-T cells still needs to be elucidated.
Adoptively transferred T cells often suffer from exhaustion and insufficient expansion, in part, because of the immunosuppressive nature of tumor-bearing hosts. To circumvent this obstacle, several combinatorial approaches, which include the immune checkpoint blockade, in vivo costimulation, and posttransfer vaccination have been developed (reviewed in Redeker and Arens60 and Beavis et al61 ). One clear advantage of CARs recognizing the peptide/MHC complex over current and conventional CARs is that there are opportunities for cross-presentation by professional APCs in draining lymph nodes and stromal cells in the tumor, which could facilitate further expansion of transferred CAR-T cells and destruction of stroma in solid tumors leading to more efficient tumor regression. In addition, posttransfer vaccination can be co-opted in CAR-T cell therapy targeting peptide/MHC, wherein the forms of antigen and its delivery are of critical importance. Synthetic LP vaccines are superior to short peptide vaccines for inducing T-cell responses (reviewed in Melief and van der Burg62 ), and we have reported recently that a synthetic LP in the form of encapsulated cholesteryl pullulan63 is efficiently taken up and cross-presented by DCs and macrophages in vitro and in vivo.64-66 Because of the technical limitation in reconstituting NOG mice with human DCs/macrophages, we could not directly show the efficacy of posttransfer vaccination to boost CAR-T cell function in vivo in the form of WT1236Y LP/CHP. Nevertheless, we showed in the present study that vaccination with human mature DCs pulsed with WT236Y, but not with an irrelevant peptide MAGE-A4143-151, enhanced regression of tumor-expressing endogenous WT1235-243 in NOG mice transferred with #213 scFv CAR-T cells. Our preliminary experiments revealed that PBMCs pulsed with WT1235-243 or synthetic LP containing WT1235-243 in the form of CHP induced only marginal proliferation of #213 scFv CAR-T cells (supplemental Figure 8), vaccination using WT1236Y, but not WT1235-243, may be warranted. The mechanisms by which DC vaccination enhances antitumor efficacy of adoptive transferred CAR-T cells recognizing tumor antigenic peptide in association with MHC appear to involve, at least partly, expansion and activation of CAR-T cells in vivo as evidenced by Ki67 and class II immunostaining, respectively, which are largely consistent with cases of adoptive transfer with TCR-based T cells.67,68 It remains to be determined where DCs present antigenic peptides to peptide/MHC specific CAR-T cells because we failed to detect human CD11c+ cells either in the tumor and the spleen (data not shown). We also found that #213 scFv CAR-T cells were effective in suppressing growth of mesothelioma, which was further enhanced by DC vaccine. This also suggests that adoptive cell therapy with #213 scFv CAR-T cells could be an important option for patients with mesothelioma who suffer from lack of satisfying therapeutics. Collectively, our present results provide a proof of concept that efficacy of CAR-T cells targeting peptide/MHC to suppress tumor growth can be boosted by vaccination. Furthermore, because vaccination with WT1236Y has been shown to induce cytotoxic T lymphocytes that could recognize and kill tumor cell with endogenous WT1 expression,35 our results raise an interesting possibility that these cytotoxic T lymphocytes may synergize with #213 scFv CAR-T cells to enhance clinical efficacy. However, a recent phase 1/2 study of WT1236Y peptide vaccine involving 26 patients resulted in 1 patient suffering an immune-related, but manageable, adverse event.69 Therefore, the possibility remains that such synergism may be associated with increased toxicity. Together with possible cross-reactivity of our #213 CAR (see the following section), a future clinical study designs will include considerations of how to mitigate such risks.
A human clinical trial of adoptive cell therapy using T cells transduced with an affinity matured TCR against MAGE-A3/HLA-A*01 resulted in fatal cardiac toxicity from off-target cross-reactivity.70 The structural nature of MHC that allows loading of a limited number of amino acids to which scFv binds may increase the likelihood of off-target cross-reactivity to self-peptides, which is an important issue to be addressed. TCRs are inferred to require approximately 5 amino acid residues on average for the recognition of a presented peptide on MHC class I.71 Our #213 CAR requires 3 amino acid residues of the WT1235 peptide for optimal recognition, which may pose a risk of cross-reactivity to self-peptides. It was indeed demonstrated that 3 risk peptides including 1 encoded by pseudogene loaded on T2A24 stimulated #213 scFv CAR-T cells to produce IFN-γ. It is mandatory to determine whether these peptides are processed and expressed on normal tissues to stimulate #213 scFv CAR-T cells to destruct normal tissues. In case of alloreactivity, #213 scFv CAR-T cells did not react to panel of LCLs expressing different, most common, HLA molecules. Our epitope prediction may still miss other epitopes recognized by #213 scFv CAR-T cells and allo-HLA cross-reactivity analysis did not involve LCLs expressing infrequent HLA molecules as well as HLA molecules presenting all tissue-specific antigens. Concerning on-target/off-tumor toxicity, expression of WT1 protein is restricted to a limited set of tissues,72,73 and the levels are considerably lower than those in leukemia and solid tumors.74,75 Although previous clinical trials of WT1 vaccine and adoptive T-cell therapy targeting WT1 have shown no on-target/off-tumor toxicity toward those healthy tissues,76,77 this might be because conventional T cells require costimulation provided by target cells when these cells express low levels of target epitopes for their optimal activation. By contrast, CAR-T cells do not require this form of costimulation and thus are more easily activated by normal cells when they express even low levels of target epitopes, which may result in on-target/off-tumor toxicity. Taken as a whole, it can’t be stressed more that rigorous toxicity testing including cross-reactivity study using peptide array that cover the substitution of all amino acids of WT1235-243 peptide with all 20 amino acids and a larger panel of LCLs expressing different allo-HLAs is needed before clinic.
In summary, we have successfully developed CAR-T cells that target an intracellular tumor-associated antigen in a HLA-A*2402–restricted manner. Because HLA-A*2402 is a subtype found predominantly in Asians (http://www.allelefrequencies.net)34 and the WT1 protein is highly expressed in both leukemia and various solid tumors, #213 scFv CAR has potentially broad targets in patients and warrants clinical trials for treatment of various tumors.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Chisaki Amaike-Hyuga, Tae Hayashi, Yuki Orito, and Koshi Sukeno for their skilled technical assistance.
This work was supported by grants from the Japan Society for the Promotion of Science Grants-in-Aid for Scientific Research Program (16K14631; Y.A.), (25670553; T.K.), (24591899; L.W.).
Authorship
Contribution: Y. Akahori, L.W., M.Y., and N.S. designed and performed experiments, analyzed results, and prepared figures; Y. Akatsuka, S. Okamoto, J.M., T.M., Y. Amaishi, and H.F. provided reagents; Y.M., S. Okumura, and H.I. provided advice for experiments; T.K. and H.S. conceived the study, interpreted data, and supervised the work; and T.K. wrote the paper.
Conflict-of-interest disclosure: Y. Amaishi, S. Okamoto, and J.M. are employees of Takara Bio Inc. which collaborated in the development of virus vector construction. The remaining authors declare no competing financial interests.
Correspondence: Takuma Kato, Department of Cellular and Molecular Immunology, Mie University Graduate School of Medicine, 2-174 Esobashi, Tsu, Mie 514-8507, Japan; e-mail: katotaku@doc.medic.mie-u.ac.jp; and Hiroshi Shiku, Department of Immuno-Gene Therapy, Mie University Graduate School of Medicine, 2-174 Esobashi, Tsu, Mie 514-8507, Japan; e-mail: shiku@clin.medic.mie-u.ac.jp.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal