

Key Points
Defined-composition manufacturing platform of CD19 CAR T cells contributes to >90% intent-to-treat complete remission rate.
Uniformity of durable persistence of CAR T cells and mitigation of antigen escape are key aspects for further optimization.
Transitioning CD19-directed chimeric antigen receptor (CAR) T cells from early-phase trials in relapsed patients to a viable therapeutic approach with predictable efficacy and low toxicity for broad application among patients with high unmet need is currently complicated by product heterogeneity resulting from transduction of undefined T-cell mixtures, variability of transgene expression, and terminal differentiation of cells at the end of culture. A phase 1 trial of 45 children and young adults with relapsed or refractory B-lineage acute lymphoblastic leukemia was conducted using a CD19 CAR product of defined CD4/CD8 composition, uniform CAR expression, and limited effector differentiation. Products meeting all defined specifications occurred in 93% of enrolled patients. The maximum tolerated dose was 106 CAR T cells per kg, and there were no deaths or instances of cerebral edema attributable to product toxicity. The overall intent-to-treat minimal residual disease–negative (MRD−) remission rate for this phase 1 study was 89%. The MRD− remission rate was 93% in patients who received a CAR T-cell product and 100% in the subset of patients who received fludarabine and cyclophosphamide lymphodepletion. Twenty-three percent of patients developed reversible severe cytokine release syndrome and/or reversible severe neurotoxicity. These data demonstrate that manufacturing a defined-composition CD19 CAR T cell identifies an optimal cell dose with highly potent antitumor activity and a tolerable adverse effect profile in a cohort of patients with an otherwise poor prognosis. This trial was registered at www.clinicaltrials.gov as #NCT02028455.
Introduction
CD19-specific chimeric antigen receptor (CAR)–expressing autologous T cells administered after lymphodepleting chemotherapy can induce clinical remissions in B-lineage malignancies, including refractory pediatric acute lymphoblastic leukemia (ALL), irrespective of disease burden or anatomic dissemination.1,,,,-6 Obstacles to broad clinical deployment of this treatment are a high failure rate in CAR T-cell manufacturing, heterogeneity of antitumor responses, and severe modality-associated toxicities in some patients.1,7,8 Although structure-function attributes of the various CD19-specific CARs have been delineated in preclinical models,9 variations among the polyclonal transduced CAR T-cell products tested in clinical trials have limited opportunities for the systematic study of product attributes related to cellular composition, differentiation, and transgene expression that affect therapeutic potency and safety.10 Additionally, a major barrier to the overall success of this therapy in other reported studies has been the exclusion of research participant enrollment in up to 24% of patients based on an in vitro assay predicting failure to manufacture a CAR T-cell product.8 Given the high rates of remission in patients with ALL, manufacturing feasibility is a significant contributor to intent-to-treat (ITT) remission induction.
We engineered a CAR T-cell product that comprises a defined 1:1 ratio of CD4+/CD8+ CAR T cells, selects for uniform high-level CAR expression, and limits activation-induced differentiation of CD4+/CD8+ T cells by using homeostatic cytokines for CAR T-cell expansion. Herein, we report a robust ITT product-manufacturing success rate of 100% in minimally selected heavily pretreated patients and a 93% minimal residual disease–negative (MRD−) complete remission (CR) rate in treated patients, resulting in 89% overall efficacy based on the ITT population, with a tolerable adverse effect profile, including an overall decrease in severity of cytokine release syndrome (CRS) compared with prior studies and no cases of cerebral edema.1,2 We report the duration of leukemic remissions and the impact of lymphodepleting regimens and CD19 antigen burden on sustained CAR engraftment and durable remissions. These data establish the feasibility of this advanced manufacturing platform and support further study of this highly defined CD19 CAR T-cell product.
Methods
Study design and participants
This phase 1/2 study for recurrent/refractory CD19+ ALL in children and young adults was conducted in accordance with US Food and Drug Administration and International Conference on Harmonisation Guidelines for Good Clinical Practice, the Declaration of Helsinki, and applicable institutional review board requirements. All patients or their guardians provided written informed consent.
Enrollment criteria included: age ≥12 months and <27 years and weight ≥10 kg. Patients with no prior history of allogeneic hematopoietic cell transplantation (HCT) were required to have one of the following characteristics: second or later marrow relapse, with or without extramedullary disease; first marrow relapse at the end of the first month of reinduction, with the marrow having ≥0.01% blasts by multiparameter flow cytometry, with or without extramedullary disease; primary refractory disease, defined as having M2 or M3 marrow after ≥2 separate induction regimens; or an indication for HCT but ineligible for the procedure. Patients who had undergone allogeneic HCT were required to have a confirmed CD19+ leukemia recurrence, defined as ≥0.01% disease, and were required to be free from active graft-versus-host disease (GVHD) and have ended immunosuppressive therapy ≥4 weeks before enrollment. Patients with central nervous system (CNS) leukemic involvement were eligible for the study, provided they were asymptomatic. Patients with significant neurologic deterioration were not eligible until alternate therapies resulted in neurologic stabilization and return to baseline status. Patients had a Lansky performance status score of ≥50 or a Karnofsky score of ≥50 for those age ≥16 years. Patients were required to have a life expectancy of ≥8 weeks and to have recovered from the acute toxic effects of prior chemotherapy, immunotherapy, or radiotherapy. Patients were required to have adequate organ function and an absolute lymphocyte count of ≥100 cells per µL.
Clinical lentiviral vector and cell product manufacture
The CD19CAR-T2A-EGFRt sequence (supplemental Figure 2, available on the Blood Web site) and expressed proteins have been previously described.11,12 CD8+ and CD4+ T cells were sequentially isolated from patient apheresis product by immunomagnetic separation using the CliniMACS device (Miltenyi Biotec) in positive selection mode. Selected cell fractions then either proceeded directly to stimulation or were cryopreserved in Cryostor-CS5 (BioLife Solutions) for future expansion.
After enrichment, 180 × 106 CD4 and CD8 selected T cells were separately stimulated with GMP Dynabeads CD3/CD28 CTS (Thermo Fisher) at a 1:3 ratio (T cell to bead) and transduced via spinoculation with clinical grade SCRI-19CAR-01_epHIV7 at an approximate multiplicity of infection of 0.1 in X-Vivo15 containing 10% fetal calf serum with 0.1 mg/mL of protamine sulfate (APP Pharmaceutical). CD4 cultures were supplemented with 5 ng/mL of recombinant human interleukin-7 (rhuIL-7) and 0.5 ng/mL of rhuIL-15 (CellGenix), whereas CD8 cultures were supplemented with 0.5 ng/mL of rhuIL-15 and 50 U/mL of rhuIL-2 (Prometheus). Cultures were then transferred to appropriately sized VueLife fluorinated ethylene propylene culture bags (Saint-Gobain) and maintained at 37°C, 75% relative humidity, 5% CO2. Cultures were fed every 2 to 3 days with fresh X-Vivo15 and 10% fetal calf serum, supplemented with appropriate cytokines as described to keep cell density between 5 × 105 and 2 × 106 viable cells per mL. Approximately 10 days after the lentiviral transduction, the CD3/CD28 CTS particles were removed using the Dynal DynaMag CTS magnet. Approximately 2 days after bead removal, cultures received immunomagnetic positive selection for EGFRt-expressing cells using the CliniMACS device.
Cultures were propagated until sufficient cells for testing and patient administration were generated as determined by Trypan blue exclusion, at which time cultures were harvested, washed in Isolyte (Braun) with 2% human serum albumin, and resuspended in Cryostor-CS5 for cryopreservation in CryoMACS bags (Miltenyi Biotec). Quality-control tests on freshly thawed cells included viability by Trypan blue exclusion, potency (cell-surface expression of EGFRt), identity (cell-surface expression of CD4 or CD8 as appropriate), average transgene copy number (WPRE quantitative polymerase chain reaction [PCR]), replication-competent virus testing (VSV-G quantitative PCR and formal replication-competent lentivirus testing at the University of Indiana), residual bead count, mycoplasma (PCR), endotoxin (limulus amebocyte lysate), and sterility (US Pharmacopeia bacterial and fungal). Typically, these release tests were completed within 7 days of product cryopreservation.
Evaluations
The primary end points of this study were to evaluate feasibility and toxicity. Feasibility was evaluated by the ability to generate a therapeutic product after 2 attempts using a single apheresis product as starting material. Toxicity was evaluated relative to a baseline assessment conducted within 24 hours before T-cell infusion. The primary efficacy end point was the MRD− rate by bone marrow aspirate by day 63; assessments were also conducted on days 7, 21, and 42 postinfusion. Responses were graded per standard ALL criteria. Secondary end points included persistence of transferred T cells (defined as the detection of transferred T cells in the peripheral blood and bone marrow), disease response and anti-CD19 activity response (defined as a measurable effect of disease reduction and absence of CD19+ cells), and efficacy of cetuximab in ablating EGFRt+ T cells. Isolated peripheral blood and bone marrow lymphocytes and whole cerebrospinal fluid were stained with a viability dye and the following monoclonal anti-human antibodies: CD3, CD4, CD8, CD19, CD14, and allophycocyanin-conjugated cetuximab (BD Biosciences). Cells were acquired on an LSRFortessa (BD, Franklin Lakes, NJ) and analyzed using FlowJo software (TreeStar, Ashland, OR). Immunophenotyping of surface markers on peripheral blood mononuclear cells and starting and final products was performed using standard staining and flow cytometry techniques with combinations of the following fluorophore-conjugated anti-human monoclonal antibodies: CD3, CD8α, CD4, CD14, CD45RO, CD27, CD45RA, CCR7, CD95, PD-1, and LAG-3 (BD Biosciences) and TIM-3 and CD39 (Biolegend). CAR T-cell expression (EGFRt) was quantified using custom-conjugated cetuximab-allophycocyanin (BD Biosciences). Cells were also stained with a live/dead viability dye (BD). Cells were acquired on an LSRFortessa (BD Biosciences), and flow cytometric analysis was performed using FlowJo software (Treestar). T cells were defined as singlets/lymphocytes/live CD3+CD14−/CD4+ or CD8+.
Statistical analysis
The maximum tolerated dose (MTD) at the end of phase 1 dose escalation was defined as the highest T-cell dose with ≥6 toxicity-evaluable patients and a cumulative dose-limiting toxicity (DLT) rate <34%.
Standard descriptive statistics (median/range, percentage, and response rate) are reported for key variables. Directional associations between categorical variables with 3 levels (severe CRS, neurotoxicity) and various predictors were evaluated via univariate proportional odds logistic regression. The significance level for these tests and others described in this section was .05.
Kaplan-Meier curves were calculated for overall survival (OS), event-free survival (EFS), and B-cell aplasia (BCA), a marker of T-cell persistence. The OS and EFS curves include data from all infused patients. For BCA, only data from patients achieving CR were included. Pairs of curves were calculated for these end points, dividing patients into 2 groups by characteristics and variables either inherent to the design or known to be risks, and log-rank tests were performed to assess significance.
Survival end points were analyzed with multivariable proportional hazards Cox models and adjusted for dose (log transformed) and CD19 burden, with incorporation before hematopoietic stem-cell transplantation (HSCT; yes or no) as a risk stratum.
For engraftment analysis, peak engraftment (as percentage of all T cells) and time-averaged engraftment (area under the curve) from infusion through day 63 were calculated for each patient, then grouped by risk factors as listed in this section, and differences were tested via a two-sided Wilcoxon-Mann-Whitney (when 2 groups were compared) or Kruskal-Wallis test (when ≥3 groups were compared).
Analyses were performed using R 3.3.1 (R Foundation for Statistical Computing, Vienna, Austria) and SAS software (version 9.4; SAS Institute, Inc., Cary, NC).
Results
Forty-five patients with relapsed or refractory CD19+ ALL were enrolled in the phase 1 arm of the PLAT-02 protocol, with a median age of 12.3 years (range, 1.3-25.4 years), including 4 patients <3 years of age (Table 1; supplemental Table 1). Twenty-eight patients (62%) had a history of at least 1 prior allogeneic transplantation, with a range of 121 to 1783 days from the time of most recent transplantation before enrollment. The disease burden varied, with 22 patients having M3 marrow and 9 having active CNS involvement at time of lymphodepletion. Seven patients had previously received CD19-directed therapy: blinatumomab (n = 6) and second-generation (CD28zeta) CD19-specific CAR T cells (n = 1). The mean time between completion of blinatumomab and PLAT-02 CAR T-cell infusion was 52 days (range, 36-73 days) and 21 months from prior CD19 CAR T-cell infusion.
Patient characteristics
| Characteristic | No. (%) of patients (N = 45) |
|---|---|
| Age, y | |
| Median | 12.2 |
| Range | 1.3-25.3 |
| Female sex | 22 (48.9) |
| Disease status | |
| Primary refractory | 3 (6.7) |
| First relapse | 15 (33.3) |
| Second relapse | 22 (48.9) |
| ≥Third relapse | 5 (11.1) |
| No. of prior allogeneic transplantations | |
| 0 | 17 (37.8) |
| 1 | 24 (53.3) |
| 2 | 4 (8.9) |
| Time since allogeneic transplantation, mo | |
| Median | 18.3 |
| Range | 4.0-58.6 |
| Disease status at lymphodepletion | |
| M3 | 22 (48.9) |
| M2 | 6 (13.3) |
| M1 | 8 (17.8) |
| MRD− | 7 (15.6) |
| CNS status at lymphodepletion | |
| CNS1 | 34 (79.1) |
| CNS2 | 7 (16.3) |
| CNS3 | 2 (4.7) |
| ALC, cells per µL | |
| Median | 1228 |
| Range | 168-4 488 |
| ABC range, cells per µL | 0-48 407 |
| Characteristic | No. (%) of patients (N = 45) |
|---|---|
| Age, y | |
| Median | 12.2 |
| Range | 1.3-25.3 |
| Female sex | 22 (48.9) |
| Disease status | |
| Primary refractory | 3 (6.7) |
| First relapse | 15 (33.3) |
| Second relapse | 22 (48.9) |
| ≥Third relapse | 5 (11.1) |
| No. of prior allogeneic transplantations | |
| 0 | 17 (37.8) |
| 1 | 24 (53.3) |
| 2 | 4 (8.9) |
| Time since allogeneic transplantation, mo | |
| Median | 18.3 |
| Range | 4.0-58.6 |
| Disease status at lymphodepletion | |
| M3 | 22 (48.9) |
| M2 | 6 (13.3) |
| M1 | 8 (17.8) |
| MRD− | 7 (15.6) |
| CNS status at lymphodepletion | |
| CNS1 | 34 (79.1) |
| CNS2 | 7 (16.3) |
| CNS3 | 2 (4.7) |
| ALC, cells per µL | |
| Median | 1228 |
| Range | 168-4 488 |
| ABC range, cells per µL | 0-48 407 |
ABC, absolute blast count; ALC, absolute lymphocyte count.
An objective of our study was to determine the feasibility of manufacturing and releasing patient-derived products with distinct features: a defined 1:1 ratio of CAR-expressing CD4+/CD8+ subsets, uniform high-level expression of CD19-specific 4-1BB:ζ CAR, and homogeneity of T-cell differentiation at the end of culture with enrichment of cells expressing cell-surface markers associated with engraftment fitness.13,14 In the heavily treated population of children and young adults with ALL enrolled in this study, the sole eligibility requirement pertaining to manufacturing feasibility was an ALC >100 cells per µL. The average ALC at the time of apheresis was 1228 cells per µL (range, 168-4488 cells), and no in vitro proliferation screen was required. All enrolled patients had successful manufacturing of a clinical CAR T-cell product, and 33 of 45 products were manufactured from fresh apheresis material, whereas 12 of 45 were manufactured from cryopreserved CD4 and CD8 T-cell subsets. Paired CD4/CD8 products were manufactured (Figure 1A) and released at the protocol-prescribed doses from 41 (89%) of 45 on an ITT basis, whereas an additional 3 patients had released products that deviated from specification (2 without CD4 unit and 1 with a less than 1:1 CD4:CD8 dose administered). One patient (S39) had a product that failed to expand adequately and required a second apheresis, which produced a CAR T-cell product that was released for infusion at the prescribed 1:1 ratio. The cytokine cocktails used for manufacturing of CD8 and CD4 T cells enriched for T cells with phenotypic markers associated with engraftment fitness, such as CD127, CCR7, and CD27 (Figure 1B). The midprocess immunomagnetic EGFRt selection step enriched for transgene-expressing T cells (Figure 1C) at a uniform level of expression (Figure 1D). The average length of cell-product manufacturing was 15 days for CD8-cell products (range, 11-22 days) and 14 days for CD4 products (range, 10-20 days). A median of 53 days transpired (range, 29-156 days) from consent to product infusion.

Product manufacturing and product phenotype. (A) Manufacturing schema of PLAT-02–formulated CD19 CAR products. (B) Flow cytometric phenotype of input apheresis T cells and final products (FPs). (C) Frequency of transgene-expressing T cells in formulated CD4 and CD8 products based on EGFRt flow cytometric enumeration. (D) EGFRt transgene expression levels of formulated products based on mean fluorescence intensity (MFI). PBMC, peripheral blood mononuclear cell; SP, starting products.
Product manufacturing and product phenotype. (A) Manufacturing schema of PLAT-02–formulated CD19 CAR products. (B) Flow cytometric phenotype of input apheresis T cells and final products (FPs). (C) Frequency of transgene-expressing T cells in formulated CD4 and CD8 products based on EGFRt flow cytometric enumeration. (D) EGFRt transgene expression levels of formulated products based on mean fluorescence intensity (MFI). PBMC, peripheral blood mononuclear cell; SP, starting products.
Detectable engraftment and expansion of CAR T cells were observed in 42 (98%) of the 43 infused patients, and BCA was observed in 40 (93%) of 43. Irrespective of cell dose, all patients (n = 14) who received prescribed lymphodepletion with fludarabine and cyclophosphamide achieved uniform engraftment of functional CAR T cells as defined by detectable CAR T cells in blood and subsequent development of BCA and MRD− remission. Engrafted CAR T cells were present in peripheral blood, marrow, and cerebrospinal fluid (Figure 2A) and were functional in each of these anatomic compartments based on ablation of malignant and nonmalignant CD19+ cells (Figure 2B). The median time to peak engraftment in peripheral blood was 10 days (range, 7-18 days). For the entire cohort of treated patients, the magnitude (area under the curve) and peak engraftment in peripheral blood did not correlate with the infused cell dose (P = .9 and .8, respectively) or disease burden at the time of product infusion (P = .12 and .14, respectively) but positively correlated with the total level of CD19+ antigen load in the marrow (tumor plus nonmalignant B cells >15% vs <15%; P = .001 and .003, respectively) and the use of fludarabine and cyclophosphamide versus cyclophosphamide alone as the lymphodepletion regimen (P = .05 and .03, respectively; Figure 2C). However, the significance of fludarabine and cyclophosphamide on peak engraftment was lost when accounting for CD19 antigen load. Engraftment kinetics in the bone marrow were similar to those in peripheral blood; however, M3 disease burden significantly correlated with the magnitude of CAR T-cell engraftment in marrow specimens (supplemental Figure 2A-B). The persistence of functional CD19 CAR T cells was assessed by measuring the duration of BCA by flow cytometry. With a median follow-up duration of 9.6 months for the entire cohort (range, 2-28 months), the median expected duration of BCA was 3 months (95% CI, 2.07-6.44; Figure 2D).

Magnitude and duration of functional CD19 CAR engraftment. (A) Representative engraftment and detection of CD19 CAR+/EGFRt+ T cells in peripheral blood (PB), bone marrow (BM), and cerebrospinal fluid (CSF) 21 days after infusion. (B) Representative functional activity of CD19 CAR product as measured by clearance of B cells (leukemic and normal) in PB, BM, and CSF. (C) Magnitude and duration of CAR engraftment by flow cytometric quantitation of EGFRt+CD3+ T cells. (D) Durability of functional CD19 CAR engraftment in treated patients based on BCA. Flu/cy, fludarabine and cyclophosphamide.
Magnitude and duration of functional CD19 CAR engraftment. (A) Representative engraftment and detection of CD19 CAR+/EGFRt+ T cells in peripheral blood (PB), bone marrow (BM), and cerebrospinal fluid (CSF) 21 days after infusion. (B) Representative functional activity of CD19 CAR product as measured by clearance of B cells (leukemic and normal) in PB, BM, and CSF. (C) Magnitude and duration of CAR engraftment by flow cytometric quantitation of EGFRt+CD3+ T cells. (D) Durability of functional CD19 CAR engraftment in treated patients based on BCA. Flu/cy, fludarabine and cyclophosphamide.
The rate of MRD− CR, as measured by multiparameter flow cytometry, was 89% (40 of 45) in all enrolled subjects and 93% (40 of 43) among research participants who received a CAR T-cell infusion (Table 2). All remissions occurred by day 21. There was no impact of disease burden, relapsed or refractory status, prior transplantation status, or high-risk cytogenetics on achievement of MRD− CR. The rate of MRD− CR was 90% (19 of 22) in M3 patients and 100% (14 of 14) for those who received fludarabine and cyclophosphamide. Four of the six patients who had received prior blinatumomab obtained an MRD− CR. Of the 3 evaluable patients who did not obtain a CR, 2 had evidence of engraftment with expansion and functional activity of the CAR T cells (supplemental Figure 3). The ALC at the time of apheresis was lower in the 3 nonresponders (mean, 469 vs 1327 cells per µL; P = .04). Of the 40 patients who obtained an MRD− CR, 27 had a malignant clone identified by next-generation sequencing. Sixty-five percent (17 of 27) of these patients achieved a molecular CR by day 63.
MRD-negative CR rate by patient subgroup
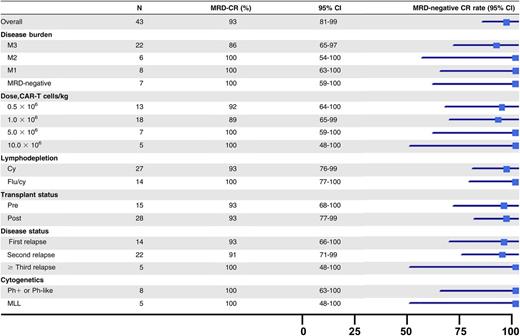 |
|
MLL, mixed lineage leukemia; Ph, Philadelphia chromosome.
The estimated 12-month EFS is 50.8% (95% CI, 36.9%-69.9%; Figure 3A) and the estimated 12-month OS is 69.5% (95% CI, 55.8%-86.5%; Figure 3B) of infused patients, with a median follow-up time of 9.6 months. The temporal relationship between duration of BCA, duration of remission, and occurrence of relapse is depicted in Figure 3C. Eighteen of the 40 patients who obtained an MRD− CR experienced relapse, 17 of which were isolated to marrow and 1 of which was isolated to CNS (supplemental Table 1). Seven relapses were associated with loss of cell-surface detection of CD19, including 1 lineage switch to acute myeloid leukemia occurring 1 month after CAR T-cell infusion.15 Of the 11 relapses in which leukemic cell-surface CD19 expression was preserved, the median time from T-cell infusion to relapse was 5.98 months (range, 1.25-14 months), with a median time from loss of BCA to relapse of 3.7 months (range, 0-11 months). Eleven patients underwent consolidative allogeneic HSCT, and 2 subsequently experienced recurrence, both with CD19+ disease. These 2 patients exhibited molecularly detectable leukemia by next-generation sequencing at the time of transplantation; 1 was MRD− by flow cytometry. Of 29 patients who reached a CR and did not proceed to consolidative allogeneic HSCT, 13 remain in continuous CR, with a median follow-up time of 12.2 months (range, 1.9-27.5 months).

Antileukemia response durability. Kaplan-Meier curves of (A) EFS and (B) OS. (C) Waterfall plot of individual research participant remission duration, BCA duration, and timing of allogeneic (allo) HSCT and/or relapse. (D) Effect of CD19 burden on BCA durability. (E) Effect of flu as component of lymphodepletion regimen on BCA durability.
Antileukemia response durability. Kaplan-Meier curves of (A) EFS and (B) OS. (C) Waterfall plot of individual research participant remission duration, BCA duration, and timing of allogeneic (allo) HSCT and/or relapse. (D) Effect of CD19 burden on BCA durability. (E) Effect of flu as component of lymphodepletion regimen on BCA durability.
In analyzing the 40 patients achieving MRD− remission in this study, we found that the loss of functional CAR T cells, as measured by loss of BCA, adversely affected the risk of CD19+ leukemic relapse (hazard ratio [HR], 34; 95% CI, 2.1-552; P = .01), whereas the HR for all relapses, CD19+ and CD19−, was 3.5 (95% CI, 1.01-11.88; P = .04). In a multivariate analysis of factors that predicted BCA duration, inclusive of dose level, disease burden, and lymphodepletion, marrow CD19+ antigen load of <15% before lymphodepletion was a significant risk factor for early loss of BCA (HR, 2.99; 95% CI, 1.32-6.81; P = .005). The median duration of BCA for patients with a CD19+ antigen load >15% was 6.4 months (95% CI, 2.6 months to upper bound not defined) vs 1.7 months for patients with a load of <15% (95% CI, 1.4 months to upper bound not defined; Figure 3D). The median duration of BCA for the 29 patients who received lymphodepletion therapy without fludarabine and cyclophosphamide was 2.1 months (95% CI, 1.4-6.4 months) versus 6.4 months (95% CI, lower bound 2.5 months) for the 14 patients who received fludarabine and cyclophosphamide (P = .15; Figure 3E).
Ten patientes received a second dose of CAR T cells (supplemental Table 2). Administration of a second dose of CAR T cells in 8 patients who had lost engraftment resulted in only 2 demonstrating evidence of reengraftment. The 6 patients who did not obtain reengraftment received the less immunosuppressive lymphodepletion regimen that did not include fludarabine with the first infusion; therefore, the role of immunologic rejection cannot be discounted as an etiology of reengraftment failure.3,4 Two (S04 and S32) of the 8 patients were administered CAR T cells for CD19+ relapse after loss of CAR T cells, with 1 (S32) obtaining a second MRD− CR. Two additional patients were administered repeat infusions for ongoing (S09) or reemergence (S37) of CD19+ disease with persistent low-level CAR T-cell engraftment; after reinfusion, these 2 patients continued to experience ongoing CAR T-cell persistence without substantial reexpansion, induction of BCA, or antileukemic effect.
The occurrence of severe toxicities resulting in patient mortality is a barrier to broad adoption of CAR T-cell immunotherapy. No deaths resulting from toxicity were reported in this trial. DLTs were observed at all dose levels and disease burdens (Table 3). Dose-level 2 (1 × 106 CAR T cells per kg) was determined to be the MTD, with a DLT rate of 17% (1 of 6) with fludarabine and cyclophosphamide and 9% (1 of 11) overall. No patient met criteria to receive cetuximab. The most common adverse events related to CAR T cells were CRS and neurotoxicity (supplemental Tables 3 and 4). The overall incidence of CRS was 93% (40 of 43) of patients, and the rate of protocol-defined severe CRS (requirement for pressors, inotropes, or respiratory failure) was 23% (10 of 43). The predominant symptom that contributed to severity of CRS was hypotension requiring pressor support. No patient required intubation for respiratory failure, multiple pressors, or high-dose pressors. For treatment of CRS, 16 patients received 1 or >1 (5 of 16) doses of tocilizumab, and 10 patients received corticosteroids for a median duration of 5 days (range, 1-13 days). The use of tociluzimab and/or corticosteroids did not affect response rates or durability of remission. A dose-level effect on severity of CRS was noted, with higher doses associated with higher rates of severe CRS (P = .03; Figure 4A). A correlation between disease burden at the time of CAR T-cell infusion with severity of CRS (P = .56) or with CD19 antigen load (P = .13) was not observed; however, there was a trend toward increased CRS with increased CD19 antigen load. Similarly, there was no impact of the lymphodepletion regimen on severity of CRS (P = .63).
Summary of DLTs
| Dose level, cells per kg | Type | Rate, n/n (%) |
|---|---|---|
| Non-flu/cy lymphodepletion | ||
| 0.5 × 106 | Grade 4 encephalopathy | 1/7 (14) |
| 1.0 × 106 | Grade 4 hydrocephalus | 1/11 (9) |
| 5.0 × 106 | — | 0/5 (0) |
| 10.0 × 106 | Grade 3 left ventricular dysfunction; | 2/5 (40) |
| Grade 3 seizure | ||
| Prescribed lymphodepletion with flu/cy | ||
| 0.5 × 106 | — | 0/6 |
| 1.0 × 106 | Grade 4 encephalopathy | 1/6 (17) |
| 5.0 × 106 | Grade 4 seizure (flu/cy); | 2/2 (100) |
| Grade 3 encephalopathy (flu/cy) |
| Dose level, cells per kg | Type | Rate, n/n (%) |
|---|---|---|
| Non-flu/cy lymphodepletion | ||
| 0.5 × 106 | Grade 4 encephalopathy | 1/7 (14) |
| 1.0 × 106 | Grade 4 hydrocephalus | 1/11 (9) |
| 5.0 × 106 | — | 0/5 (0) |
| 10.0 × 106 | Grade 3 left ventricular dysfunction; | 2/5 (40) |
| Grade 3 seizure | ||
| Prescribed lymphodepletion with flu/cy | ||
| 0.5 × 106 | — | 0/6 |
| 1.0 × 106 | Grade 4 encephalopathy | 1/6 (17) |
| 5.0 × 106 | Grade 4 seizure (flu/cy); | 2/2 (100) |
| Grade 3 encephalopathy (flu/cy) |

Severity of cytokine release syndrome and neurotoxicity. (A) Severity of CRS in treated patients as a function of cell dose, disease burden, CD19 burden, and lymphodepletion regimen. (B) Severity of neurotoxicity based on parameters described in (A) as well as severity of CRS.
Severity of cytokine release syndrome and neurotoxicity. (A) Severity of CRS in treated patients as a function of cell dose, disease burden, CD19 burden, and lymphodepletion regimen. (B) Severity of neurotoxicity based on parameters described in (A) as well as severity of CRS.
The overall incidence of neurotoxicity was 49% (21 of 43); severe neurotoxicity (any grade seizure or grade 3/4 neurotoxicity exclusive of headache) occurred in 21% (9 of 43). No cases of cerebral edema were encountered. There was no effect of dose level (P = .32), disease burden (P = .93), CD19 antigen load (P = .23), or fludarabine and cyclophosphamide lymphodepletion (P = .32) on the occurrence of severe neurotoxicity; however, the presence of severe CRS was predictive of subsequent severe neurotoxicity (P = .01; Figure 4B). All patients experiencing neurotoxicity experienced reversible symptoms and eventual return to baseline neurologic status.
Twenty-seven patients in the trial had undergone prior allogeneic HSCT. One of these patients experienced recrudescence of grade 3 acute skin GVHD (supplemental Figure 4). This patient (S03) was 2.3 years from prior transplantation and had tapered off GVHD medications more than 1 year before CAR T-cell treatment. Although the infused CAR T cells cannot be formally ruled out as direct mediators of the skin GVHD, a biopsy of involved skin revealed that 9% of CD3+ T cells infiltrating the dermis were EGFRt+ compared with 78% EGFRt+ T cells in peripheral blood. The patient was treated with prednisone 2 mg/kg per day followed by a taper for approximately 1 month, with resolution of GVHD and without untoward effect on the persistence of CAR T cells or BCA. Functional persistence of CAR T cells was noted until the time of CD19− relapse 8 months after CAR T-cell infusion.
Discussion
This study evaluated treatment of children and young adults with relapsed or refractory B-lineage ALL with CD19 CAR T cells characterized by a defined CD4/CD8 T-cell ratio, a uniform level of CAR expression, and a less differentiated phenotype. Of 43 patients treated at CAR T-cell doses ranging from 0.5 to 10 × 106 cells per kg, 41 (93%) of 43 obtained an MRD− CR, with a rate of 89% among all enrolled patients. After fludarabine and cyclophosphamide lymphodepletion, 14 of 14 patients experienced MRD− remission. Specific subgroups of patients that may predict for increased or decreased efficacy were not identified, because of the high CR rate and trial size. In particular, there was no difference in response rates based on number of prior relapses or among low and high disease burden patients. The estimated 12-month OS for this poor-prognosis cohort of patients is 65.9%, and the estimated 12-month EFS is 50.3%. A longer duration of postremission functional persistence of CAR T cells, as inferred by ongoing BCA, correlated significantly with the durability of remission. Higher CD19 antigen load positively correlated with prolonged CAR T-cell persistence and BCA.
There is significant heterogeneity in CD19 CAR clinical trials reported to date with respect to receptor composition and product formulation.1,2,4,7,16,17 Accordingly, early-phase trials have reported a spectrum of efficacy and toxicity results. Our data show that, despite significant interpatient heterogeneity of apheresis T-cell populations from children and young adults with relapsed or refractory ALL, manufacturing using cell purification, enrichment of transgene expression, and cocktails of recombinant human cytokines can reproducibly generate products that exhibit phenotypic and functional attributes associated with therapeutic potency.13,14 This is in contrast to reported studies that required a screening in vitro proliferation assay or ALC of >500 cells per µL for enrollment and excluded 24% of patients enrolled in CD19 CAR trials.8 The cell selection, homeostatic cytokines, and enrichment of transgene-expressing T cells likely all contributed to the robustness of our product-manufacturing approach. Given the efficacy of CD19 CAR T-cell infusions in inducing leukemic remissions in select trials, the greatest ITT risk of therapeutic failure now resides in the feasibility of manufacturing a product for any given research participant. This manufacturing platform therefore provides a significant advantage over prior reported trials, which excluded substantial numbers of patients because of an inability to manufacture a product or had a marked decrease in efficacy.1,2
No deaths resulting from toxicity occurred in this trial, with an acceptable incidence of severe CRS (23%) and severe neurotoxicity (23%).1,-3,7 The rate of severe CRS increased with cell dose-level escalation; however, we did not find an effect of higher disease burden with increased rates of severe CRS. The level of CD19 antigen burden, although not statistically significant, did trend toward higher rates of severe CRS. Although the reported rate of severe CRS was 23%, the threshold definition of severe CRS used for this trial was lower than that in other reported comparable trials.1,2 When applying the definition of severe CRS from other protocols, the rate of severe CRS is remarkably lower, and in fact, there were no patients meeting the definition of grade 4 severe CRS from other trials.1 Therefore, this product seems to have an improved toxicity profile, with decreased severity of CRS. It has previously been shown in animal models that the defined-composition product can induce remissions at lower doses,10 and we believe that the defined-composition formulation of this product enables the use of lower doses of CAR T cells, while retaining high efficacy, resulting in attenuated CRS severity.
This phase 1 study investigated tolerability of formulated CAR T-cell product infusions over a range of cell doses and with various lymphodepletion chemotherapy regimens. At the MTD of 106 cells per kg after fludarabine and cyclophosphamide lymphodepletion, we observed uniform engraftment and MRD− remissions in 14 of 14 research participants and enhanced durability of functional CAR T-cell persistence. Our phase 1 trial has revealed that a subset of patients exhibit durable remissions without consolidative allogeneic HSCT, and the durability of remission is enhanced by ongoing CAR T-cell persistence. A risk factor for relapse with CD19+ disease is a short duration of BCA that in turn can be predicted by the quantity of CD19-expressing B cells (malignant and nonmalignant) in the bone marrow before CAR T-cell dosing. Our observation that low quantities of CD19+ B cells result in suboptimal expansion and persistence of CD19 CAR T cells indicates a future challenge when this modality is applied among patients earlier in the course of their therapy, for instance, patients in second remission as a consolidative therapy that obviates prolonged chemotherapy and/or allogeneic HSCT. Approaches to augment CD19 CAR T-cell persistence independent of B-cell/leukemic cell burden are warranted and are being pursued by our group via the provision of T cells transduced to express CD19 after CAR T-cell infusion. Additionally, the uniform use of fludarabine and cyclophosphamide lymphodepletion may also promote longer persistence of the CAR T cells, allowing more comparable functional CAR T-cell persistence to other CD19 CAR T cells with 4-1BB costimulation.1 Although we did not observe a statistically significant advantage in the duration of CAR T-cell persistence with fludarabine and cyclophosphamide lymphodepletion, there was a trend toward improved persistence, and this has previously been shown by others to significantly enhance long-term CAR T-cell persistence, potentially through the reduction of T-cell rejection responses to the murine scFv CAR binder domain.3,4 For this reason, a phase 2 arm of this protocol is now enrolling and treating patients at the MTD of 106 cells per kg after fludarabine and cyclophosphamide lymphodepletion.
CD19 antigen escape is an established mechanism of relapse after remissions induced by CD19 CAR T-cell immunotherapy.18 In this phase 1 study, relapse occurred in 45% of patients who achieved an MRD− CR, and CD19-negative escape variant leukemia accounted for 39% of these relapses. Targeting B-lineage ALL with CD22-specific CAR T cells may enable the salvage of a proportion of patients who experience a CD19− relapse.19 Current efforts by our group are focusing on bispecific CAR engineering and vector technologies that allow for expression of 2 CARs that enable dual targeting of CD19 and CD22, because the simultaneous targeting of >2 antigens is anticipated to reduce therapeutic failures resulting from antigen escape after immunotherapy with monospecific CAR T cells.
The full-text version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
Medical writing and graphical illustration support were provided by Joe Petroziello and Olivia Lee, both employees of Juno Therapeutics, and by MediTech Media.
This work was supported in part by a Stand Up to Cancer and St. Baldrick’s Pediatric Dream Team Translational Research Grant (SU2C-AACR-DT1113), National Institutes of Health, National Cancer Institute Grant RO1 CA136551-05, an Alex’s Lemonade Stand Phase I/II Infrastructure Grant, a Conquer Cancer Foundation Career Development Award, the Washington State Life Sciences Discovery Fund, the Ben Towne Foundation, the William Lawrence and Blanche Hughes Foundation, and Juno Therapeutics, Inc.
Authorship
Contribution: R.A.G., J.R.P., and M.C.J. designed, conducted, and analyzed the study and wrote the paper; D.L. and A.P.O. analyzed the data and wrote the paper; M.B. designed and reviewed the paper; C.A., K.L., and C.S. conducted the study and reviewed the paper; O.F. and H.B. designed, conducted, and analyzed the study and reviewed the paper; C.L., S.M., and C.B. manufactured the products; K.S.K.-S. designed the study and reviewed the paper; V.H. conducted the study; and S.R.R. designed the study and reviewed the paper.
Conflict-of-interest disclosure: D.L. is an employee of and has an equity interest in Juno Therapeutics, Inc. S.R.R. has received consulting fees and grants from and is an inventor of patents licensed to Juno Therapeutics, Inc., in which he has an equity interest. M.C.J. has received consulting fees and grants from and is an inventor of patents licensed to Juno Therapeutics, Inc., in which he has an equity interest. Seattle Children’s Hospital received funds from Juno Therapeutics, Inc. The remaining authors declare no competing financial interests.
Correspondence: Michael C. Jensen, Ben Towne Center for Childhood Cancer Research, Seattle Children’s Research Institute, 1100 Olive Way, Suite 100, Seattle, WA 98101; e-mail: michael.jensen@seattlechildrens.org.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal