

Key Points
WAS patients and WAS knockout mice have fewer Tfh cells, but they express higher levels of ICOS than controls.
WASp is involved in the development and memory response of Tfh cells, likely through the regulation of the transcription factor BCL6.
Abstract
Wiskott-Aldrich syndrome protein (WASp) is a hematopoietic-specific regulator of actin nucleation. Wiskott-Aldrich syndrome (WAS) patients show immunodeficiencies, most of which have been attributed to defective T-cell functions. T follicular helper (Tfh) cells are the major CD4+ T-cell subset with specialized B-cell helper capabilities. Aberrant Tfh cells activities are involved in immunopathologies such as autoimmunity, immunodeficiencies, and lymphomas. We found that in WAS patients, the number of circulating Tfh cells was significantly reduced due to reduced proliferation and increased apoptosis, and Tfh cells were Th2 and Th17 polarized. The expression of inducible costimulator (ICOS) in circulating Tfh cells was higher in WAS patients than in controls. BCL6 expression was decreased in total CD4+ T and Tfh cells of WAS patients. Mirroring the results in patients, the frequency of Tfh cells in WAS knockout (KO) mice was decreased, as was the frequency of BCL6+ Tfh cells, but the frequency of ICOS+ Tfh cells was increased. Using WAS chimera mice, we found that the number of ICOS+ Tfh cells was decreased in WAS chimera mice, indicating that the increase in ICOS+ Tfh cells in WAS KO mice was cell extrinsic. The data from in vivo CD4+ naive T-cell adoptive transfer mice as well as in vitro coculture of naive B and Tfh cells showed that the defective function of WASp-deficient Tfh cells was T-cell intrinsic. Consistent findings in both WAS patients and WAS KO mice suggested an essential role for WASp in the development and memory response of Tfh cells and that WASp deficiency causes a deficient differentiation defect in Tfh cells by downregulating the transcription level of BCL6.
Introduction
Wiskott-Aldrich syndrome (WAS) is a rare X-linked immunodeficiency characterized by combined immunodeficiency, congenital thrombocytopenia, eczema, and an increased risk of autoimmune diseases and lymphoid malignancies.1 The disease is caused by mutations in the gene WAS, encoding the Wiskott-Aldrich syndrome protein (WASp). WASp is a key regulator of the actin cytoskeleton via activation of the Arp2/3 complex.2 WASp expression is absent in peripheral blood mononuclear cells (PBMCs) of patients with the classical WAS phenotype.
WASp deficiency results in a wide range of defects in immune cells, causing a range of immunological symptoms. Immune deficiency is the most prominent symptom and has been suggested to be mainly attributable to defective T-cell function. T helper 1 (Th1) cells from WAS patients show defective cytokine secretion, probably due to a decreased expression of T-bet messenger RNA (mRNA) and an inability to translocate NFAT1/2 to the nucleus.3,4 The secretion of Th2 cytokine by WAS−/− CD4+ T cells is also significantly reduced, although they are still able to upregulate the mRNA level of GATA3 after anti-CD3 restimulation.5 A recent study reported an increase in Th17 cells in WAS knockout (KO) mice, which was associated with exacerbated arthritis.6 However, T follicular helper (Tfh) cells, a CD4+ T-cell subset critical for B-cell differentiation,7 have not been examined in WAS patients or WAS KO mice.
Tfh cells express the chemokine receptor 5 (CXCR5), which allows them to migrate into B-cell follicles.8,9 Tfh cells also express the costimulatory molecule inducible costimulator (ICOS), CD40 ligand (CD40L), and programmed cell death 1 (PD-1) and secrete the cytokine interleukin-21 (IL-21), all of which play important roles in Tfh-cell differentiation and the development of germinal centers (GCs).7 The transcription factor BCL6 is a master regulator of Tfh-cell differentiation and function,10 whereas BLIMP1 suppresses BCL6 function.11
In humans, Tfh cells are mostly located in the light zone of the GC in secondary lymph nodules.7 CXCR5+CD4+ T cells in the peripheral blood have been identified as Tfh-like cells, exhibiting the same B-cell helper qualities as memory Tfh cells that have passed through a GC reaction.12 Approximately 20% of human central memory CD4+ T cells are CXCR5+, demonstrating that memory Tfh cells are a major component of human T-cell memory.13 We have previously reported that T-cell receptor (TCR) repertoire development and expansion of memory CD4+ T cells in WAS patients are impaired.14 At the cellular level, WASp is required for the formation of immunological synapse and TCR-mediated activation in CD4+ T cells. The stability of the synapse formed between T cells and dendritic cells is essential for costimulatory receptor engagement and/or cytokine exposure and thereby Tfh-cell differentiation.15,16 Given the known defects in WASp-deficient CD4+ T lymphocytes, we hypothesized that WASp deficiency may impair the differentiation and function of Tfh, contributing to the immunodeficiency in WAS.
In this study, we determined the number and key features of circulating Tfh cells in patients with WAS and in WAS KO mice after secondary immunization. Our results suggest that WASp plays a critical role in the generation of Tfh cells and Tfh-mediated memory response and that WASp-deficient Tfh cells contribute to the pathogenesis of immunodeficiency and autoimmunity in WAS.
Materials and methods
WAS patients and control subjects
Blood samples were obtained from patients with WAS mutations and healthy control (HC) subjects. The diagnosis of WAS was made based on clinical signs and symptoms, WAS mutations, and WASp expression measured by flow cytometric analysis as described before.17,18 Examples of different types of WASp expression are shown in supplemental Figure 1 (available on the Blood Web site). This study was conducted in accordance with the tenets of the Declaration of Helsinki and was approved by the ethics committee of Chongqing Medical University.
Adoptive transfer
Naive CD4+ T cells from spleen of WAS KO or wild-type (WT) mice were sorted by a cell sorter (Miltenyi Biotec) and injected into CD4 KO mice (4 × 106/per mouse). The recipient mice were immunized with hapten 4-hydroxy-3-nitrophenylacetyl (NP) coupled to the carrier keyhole limpet hemocyanin (KLH) the next day and analyzed at day 7 postimmunization.
Tfh-cell and B-cell coculture
Naive B cells were cocultured with Tfh cells (2 × 104 cells per well) in the presence of endotoxin-reduced staphylococcal enterotoxin B (SEB) (1 mg/mL) in RPMI 1640 complete medium supplemented with 10% fetal bovine serum. Culture medium was collected at day 3, and the levels of immunoglobulin G (IgG) and IgM were determined by enzyme-linked immunosorbent assay (ELISA) according to the manufacturer’s instructions (Bethyl Laboratories).
Statistical analyses
All statistics were performed using SPSS version 22.0 software. The data are presented as the mean and standard deviation. The significance of the difference was analyzed using a Student t test or nonparametric test (Mann-Whitney U test). P < .05 was considered statistically significant.
Results
Decreased frequency of CXCR5+CD4+ T cells in peripheral blood of WAS patients
This study included a total of 35 patients and 35 HCs. As WAS is an early-onset disease, most patients in this study were infants. The median age at sample collection for both WAS patients (0.25-10 years) and HCs (0.25-7 years) was 1 year, and the difference in age between groups was not statistically significant. The clinical characteristics of the patients are shown in supplemental Table 1. To determine whether WASp plays any role in Tfh cells, we first analyzed the frequency of CXCR5+CD4+ T cells in the peripheral blood from WAS patients and HCs. We found that WAS patients displayed a lower frequency and number of CXCR5+ cells within total CD4+ T cells than HCs (Figure 1A-B). However, the percentage of CD45RA− memory CD4+ T cells in WAS patients was significantly higher (Figure 1C), suggesting that the decrease in CXCR5+CD4+ T cells is not simply caused by a reduction in total CD4+ T cells (Figure 1D). WAS patients also showed greater reductions in CXCR5 expression on CD4+ T cells with age (Figure 1E). Furthermore, the frequency and number of CXCR5+ cells within CD45RA− memory CD4+ T cells were considerably reduced across all ages in WAS patients (Figure 1F-G). We then investigated whether the emergence of autoimmunity influenced the frequency of CXCR5+ cells in WAS using patients scored as 5A. The frequency of CXCR5+CD4+ cells in 5A WAS patients was also significantly decreased compared with HCs (Figure 1H). Furthermore, all patients with negative WASp expression clearly showed a decreased frequency of CXCR5+CD4+ cells in blood (Figure 1I). Taken together, our data indicate that Tfh cells in WAS patients are deficient due to reduced numbers, which appears to be associated with the severity of clinical manifestation and the level of WASp expression.
![Figure 1. WAS patients have fewer CXCR5+ CD4+ T cells in blood. (A) Flow cytometric analysis of CD4+CXCR5+ T cells in WAS patients and HCs. (B) Percentage and number of CXCR5+ cells within total CD4+ T cells in WAS patients (n = 35) and HCs (n = 35). (C) Percentage of CD45RA− memory cells within total CD4+ T cells in WAS patients (n = 24; patient 1 [P1]-P24) and HCs (n = 24). (D) Percentage and number of CD4+ T cells in WAS patients (n = 35) and HCs (n = 35). (E) Percentage of CXCR5+ cells within total CD4+ T cells at different ages. (F) Percentage of CXCR5+ cells within CD45RA− memory CD4+ T cells in WAS patients (n = 24; P1-P24) and HCs (n = 24). (G) Percentage of CXCR5+ cells within CD45RA− memory CD4+ T cells at different ages. Linear regression analyses in HCs or WAS patients are indicated by dotted lines and solid lines, respectively. The 95% confidence interval for each is indicated by the neighboring thinner dotted lines. (H) Percentages of CXCR5+ cells within CD4+ T cells in WAS patients with clinical scores of 3 to 4, 5A and age-matched HCs. (I) Percentages of CXCR5+ cells within CD4+ T cells in WASp-positive (WASP+), WASp-negative (WASP−), and WASp double-peak patients (WASP-DP) and age-matched HCs.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/127/25/10.1182_blood-2015-06-652636/5/m_3180f1.png?Expires=1763595712&Signature=f2riwKyn-f78TLfBv5o11Qkc3ZBz3IZmD28FWf6OCZHtvckXtvwLzJqk8N6UVhQEKdoKtuheuriTQB8~NCkVruTqLD3X~fLi3gVeQlIRhKFEYbU-~mLLUxQSAGKjcGk34tvOnfoxm6p3R1PVWhFXeROeR3R6ks1cp68G5Ne9NySwReb~u5qfLxUKr8TT4UjNV73lCzL4YmRclq0ONhSmTJO7lhgRWbynwMAUHxkhaf1Aq8TVr2ptUUkfQ67bVcPOVCJtqn-qUXgpNf3593~Yattz8ASKyXrzfFnvgOfpSRkxiwlFYScwBdg~Xcg6smDrOjCo808pKmg-KxYhj50reg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
WAS patients have fewer CXCR5+ CD4+ T cells in blood. (A) Flow cytometric analysis of CD4+CXCR5+ T cells in WAS patients and HCs. (B) Percentage and number of CXCR5+ cells within total CD4+ T cells in WAS patients (n = 35) and HCs (n = 35). (C) Percentage of CD45RA− memory cells within total CD4+ T cells in WAS patients (n = 24; patient 1 [P1]-P24) and HCs (n = 24). (D) Percentage and number of CD4+ T cells in WAS patients (n = 35) and HCs (n = 35). (E) Percentage of CXCR5+ cells within total CD4+ T cells at different ages. (F) Percentage of CXCR5+ cells within CD45RA− memory CD4+ T cells in WAS patients (n = 24; P1-P24) and HCs (n = 24). (G) Percentage of CXCR5+ cells within CD45RA− memory CD4+ T cells at different ages. Linear regression analyses in HCs or WAS patients are indicated by dotted lines and solid lines, respectively. The 95% confidence interval for each is indicated by the neighboring thinner dotted lines. (H) Percentages of CXCR5+ cells within CD4+ T cells in WAS patients with clinical scores of 3 to 4, 5A and age-matched HCs. (I) Percentages of CXCR5+ cells within CD4+ T cells in WASp-positive (WASP+), WASp-negative (WASP−), and WASp double-peak patients (WASP-DP) and age-matched HCs.
WAS patients have fewer CXCR5+ CD4+ T cells in blood. (A) Flow cytometric analysis of CD4+CXCR5+ T cells in WAS patients and HCs. (B) Percentage and number of CXCR5+ cells within total CD4+ T cells in WAS patients (n = 35) and HCs (n = 35). (C) Percentage of CD45RA− memory cells within total CD4+ T cells in WAS patients (n = 24; patient 1 [P1]-P24) and HCs (n = 24). (D) Percentage and number of CD4+ T cells in WAS patients (n = 35) and HCs (n = 35). (E) Percentage of CXCR5+ cells within total CD4+ T cells at different ages. (F) Percentage of CXCR5+ cells within CD45RA− memory CD4+ T cells in WAS patients (n = 24; P1-P24) and HCs (n = 24). (G) Percentage of CXCR5+ cells within CD45RA− memory CD4+ T cells at different ages. Linear regression analyses in HCs or WAS patients are indicated by dotted lines and solid lines, respectively. The 95% confidence interval for each is indicated by the neighboring thinner dotted lines. (H) Percentages of CXCR5+ cells within CD4+ T cells in WAS patients with clinical scores of 3 to 4, 5A and age-matched HCs. (I) Percentages of CXCR5+ cells within CD4+ T cells in WASp-positive (WASP+), WASp-negative (WASP−), and WASp double-peak patients (WASP-DP) and age-matched HCs.
Circulating Tfh subsets are altered in WAS patients
CD4+CXCR5+ T cells can be classified into Th1-like (CXCR3+CCR6−), Th2-like (CXCR3−CCR6−), and Th17-like (CXCR3−CCR6+) subsets according to the expression of CXCR3 and CCR6.19 The Th2-like and Th17-like Tfh-cell subsets have previously been shown to help B cells more efficiently than the Th1-like Tfh-cell subset.16 To determine if WASp deficiency affects the Tfh subsets differentially, we analyzed the frequency of Tfh subsets in PBMCs from WAS patients and HCs. The frequency of Th1-like Tfh cells within memory CXCR5+CD4+ T cells was significantly lower in WAS patients (particularly in those lacking WASp expression with WAS clinical scores of 3-4, 5A) than in HCs (Figure 2A-D). In contrast, the frequencies of Th2-like and Th17-like Tfh cells within CXCR5+CD4+ T cells were higher among WAS patients with clinical scores of 3-4, 5A than in HCs (Figure 2A-D). The ratio of Th2-like + Th17-like to Th1-like Tfh cells showed an enhanced polarization of Tfh subsets to Th2/Th17-like cells, the Tfh subsets that were more efficient B-cell helpers than Th1-like cells, in WAS patients with a score of 5A and no WASp expression (Figure 2E-G).
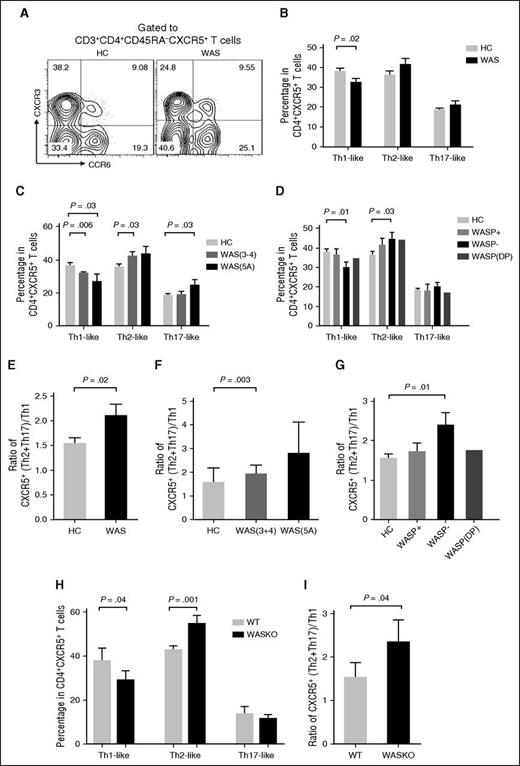
Circulating Tfh subsets are altered in WAS patients. (A) Flow cytometric analysis of CD4+CXCR5+ T-cell subsets in WAS patients and HCs. (B) Percentage of CD4+CXCR5+ T-cell subsets in WAS patients (n = 24; P1-P24) and HCs (n = 24). (C) Percentage of CD4+CXCR5+ T-cell subsets in WAS patients with clinical scores of 3 to 4, 5A and age-matched HCs. (D) Percentage of CD4+CXCR5+ T-cell subsets in WASp-positive, WASp-negative, and WASp double-peak patients and age-matched HCs. (E) Ratio of CXCR5+ (Th2 + Th17)/Th1 cells. (F) Ratio of CXCR5+(Th2 + Th17)/Th1 cells in WAS patients with clinical scores of 3 to 4, 5A and age-matched HCs. (G) Ratio of CXCR5+(Th2 + Th17)/Th1 cells in WASp-positive (WASP+), WASp-negative (WASp−), and WASp double-peak patients (WASp-DP) and age-matched HCs. (H) Percentages of Th1-like, Th2-like, and Th17-like Tfh cells from spleen of WT and WAS KO mice. (I) Ratio of CXCR5+(Th2 + Th17)/Th1 cells from spleen of WT and WAS KO mice.
Circulating Tfh subsets are altered in WAS patients. (A) Flow cytometric analysis of CD4+CXCR5+ T-cell subsets in WAS patients and HCs. (B) Percentage of CD4+CXCR5+ T-cell subsets in WAS patients (n = 24; P1-P24) and HCs (n = 24). (C) Percentage of CD4+CXCR5+ T-cell subsets in WAS patients with clinical scores of 3 to 4, 5A and age-matched HCs. (D) Percentage of CD4+CXCR5+ T-cell subsets in WASp-positive, WASp-negative, and WASp double-peak patients and age-matched HCs. (E) Ratio of CXCR5+ (Th2 + Th17)/Th1 cells. (F) Ratio of CXCR5+(Th2 + Th17)/Th1 cells in WAS patients with clinical scores of 3 to 4, 5A and age-matched HCs. (G) Ratio of CXCR5+(Th2 + Th17)/Th1 cells in WASp-positive (WASP+), WASp-negative (WASp−), and WASp double-peak patients (WASp-DP) and age-matched HCs. (H) Percentages of Th1-like, Th2-like, and Th17-like Tfh cells from spleen of WT and WAS KO mice. (I) Ratio of CXCR5+(Th2 + Th17)/Th1 cells from spleen of WT and WAS KO mice.
We next examined the production of signature cytokines by different T-cell subsets induced by in vitro stimulation by measuring the percentage of cytokine-producing CD4+CXCR5+ Tfh cells. In contrast to the reduced percentage of Th1-like Tfh cells, the percentage of interferon-γ–producing Tfh cells was increased in WAS patients compared with HCs (Figure 2B; supplemental Figure 2A). The number of IL-4–producing Tfh cells did not change, and the number of IL-17–producing Tfh cells was higher in WAS patients than in HCs (supplemental Figure 2A). Using the WAS KO mouse model, we found that the percentage of interferon-γ–secreting cells in splenic Tfh cells increased but the percentages of IL-4 and IL-17–secreting Tfh cells were unchanged in WAS KO mice compared with WT mice (Figure 2H and supplemental Figure 2B). The ratio of Th2-like + Th17-like to Th1-like Tfh cells was also increased in WAS KO mice (Figure 2I). Our results show that WASp deficiency leads to altered Tfh-cell subsets in human blood.
CD4+CXCR5+ T cells in WAS patients display higher levels of ICOS expression
ICOS signaling is required for the generation of Tfh cells.12 To investigate the mechanism leading to the reduced number of CXCR5+ CD4+ T cells in WAS patients, we examined whether WASp deficiency has an impact on ICOS costimulation. We found that WAS patients had a higher frequency of ICOS+ cells within CXCR5+CD4+ T cells than HCs (Figure 3A-B). Because the expression level of ICOS is elevated in activated T cells, we analyzed CXCR5+CD4+ T cells after in vitro stimulation with phytohaemagglutinin (PHA) for 3 days. After activation, the frequency of ICOS+ cells within CXCR5+CD4+ T cells from WAS patients was significantly higher (Figure 3A,C), indicating that the level of ICOS expression was increased in a higher number of Tfh cells in WAS patients. Using immunofluorescence microscopy, we further determined the subcellular location and the mean fluorescence intensity (MFI) of ICOS in Tfh cells. We found the MFI of ICOS in Tfh cells of WAS patients was increased on the plasma membrane and in the cytoplasm compared with HCs (Figure 3D-E). Although ICOS staining was primarily concentrated at the cell surface of Tfh cells in HCs, it appeared to be on the plasma membrane as well as in the cytoplasm (Figure 3D-E). However, there was a fair amount of ICOS staining in the cytoplasm of Tfh cells in WAS patients (Figure 3D-E). These results suggested that WASp deficiency caused an increase in the expression levels of ICOS in Tfh cells.

WAS patients display more ICOS expression in CD4+ CXCR5+ T cells. (A) Flow cytometric analysis of ICOS+ cells in whole blood and stimulated PBMCs from WAS patients and HCs. (B) Percentage of ICOS+ cells within CD4+CXCR5+ T cells in WAS patients (n = 18; P1-P18) and HCs (n = 18). (C) Percentage of ICOS+ cells within CD4+CXCR5+ T cells after stimulation with PHA for 3 days in WAS patients (n = 8; P1-P8) and HCs (n = 7). (D) Representative images of ICOS staining in Tfh cells from HCs and WAS patients. Scale bar, 2.5 μm. (E) MFI of ICOS on the plasma membrane and cytoplasm of Tfh cells from HCs and WAS patients.
WAS patients display more ICOS expression in CD4+ CXCR5+ T cells. (A) Flow cytometric analysis of ICOS+ cells in whole blood and stimulated PBMCs from WAS patients and HCs. (B) Percentage of ICOS+ cells within CD4+CXCR5+ T cells in WAS patients (n = 18; P1-P18) and HCs (n = 18). (C) Percentage of ICOS+ cells within CD4+CXCR5+ T cells after stimulation with PHA for 3 days in WAS patients (n = 8; P1-P8) and HCs (n = 7). (D) Representative images of ICOS staining in Tfh cells from HCs and WAS patients. Scale bar, 2.5 μm. (E) MFI of ICOS on the plasma membrane and cytoplasm of Tfh cells from HCs and WAS patients.
CD4+ T and Tfh cells in WAS patients show normal expression of IL-21 and BLIMP1 but reduced expression of BCL6
To determine whether the cytokine secretion of circulating Tfh cells was skewed in WAS patients, both CD4+ T cells and CD4+CXCR5+ T cells from WAS patients and HCs were analyzed for the expression of IL-21. The frequency of IL-21+ cells within CD4+CXCR5+ T cells was not substantially different between WAS patients and HCs (Figure 4A-B). IL-21 mRNA expression was analyzed in sorted CD4+ T cells from patients and HCs, and again no significant difference was found (Figure 4C). We further quantified the gene expression levels of BCL6 and PRDM1 (a gene encoding the BLIMP1 protein) to determine whether the reduced number of CXCR5+CD4+ cells is associated with abnormal expression of these transcription factors. There was a marked decrease in BCL6 mRNA expression in CD4+ T cells of WAS patients compared with HCs (Figure 4D). The expression of PRDM1 mRNA in CD4+ T cells was not significantly different between WAS patients and HCs (Figure 4E). Similarly, the mRNA levels of BCL6, but not IL-21 or PRDM1, in sorted Tfh cells was significantly decreased in WAS patients compared with HCs (Figure 4F-H). These data indicate that WASp deficiency suppresses BCL6 expression.

Cytokine secretion and expression of transcription factors in WAS. (A) Flow cytometric analysis of IL-21+ cells in WAS patients and HCs. (B) Percentage of IL-21+ cells within CD4+CXCR5+ T cells in WAS patients (n = 11; P1-P11) and HCs (n = 15). (C) Gene expression of IL-21 mRNA of CD4+ T cells in WAS patients (n = 5; P1, P6-P9) and HCs (n = 6). (D) Gene expression of BCL6 mRNA of CD4+ T cells in WAS patients (n = 5) and HCs (n = 6). (E) Gene expression of PRDM1 mRNA of CD4+ T cells in WAS patients (n = 5) and HCs (n = 6). (F) Gene expression of IL-21 mRNA of Tfh cells in WAS patients and HCs (n = 6). (G) Gene expression of BCL6 mRNA of Tfh cells in WAS patients and HCs (n = 6). (H) Gene expression of PRDM1 mRNA of Tfh cells in WAS patients and HCs (n = 6).
Cytokine secretion and expression of transcription factors in WAS. (A) Flow cytometric analysis of IL-21+ cells in WAS patients and HCs. (B) Percentage of IL-21+ cells within CD4+CXCR5+ T cells in WAS patients (n = 11; P1-P11) and HCs (n = 15). (C) Gene expression of IL-21 mRNA of CD4+ T cells in WAS patients (n = 5; P1, P6-P9) and HCs (n = 6). (D) Gene expression of BCL6 mRNA of CD4+ T cells in WAS patients (n = 5) and HCs (n = 6). (E) Gene expression of PRDM1 mRNA of CD4+ T cells in WAS patients (n = 5) and HCs (n = 6). (F) Gene expression of IL-21 mRNA of Tfh cells in WAS patients and HCs (n = 6). (G) Gene expression of BCL6 mRNA of Tfh cells in WAS patients and HCs (n = 6). (H) Gene expression of PRDM1 mRNA of Tfh cells in WAS patients and HCs (n = 6).
Blood and spleen of immunized WAS KO mice contain fewer CXCR5+CD4+ T cells
We used a WAS KO mouse model to verify the observations made in WAS patients. In addition to Tfh cells, T follicular regulatory (Tfr) cells are a newly defined subset of CXCR5+CD4+ T cells. In addition to expressing high levels of CXCR5, ICOS, and PD-1 as Tfh cells, Tfr cells express Foxp3 and exert a repressive function.20-22 Here, we used Foxp3 to distinguish Tfh from Tfr cells (supplemental Figure 3). In the absence of immunization, we found the frequency and number of CXCR5+ T cells within both splenic and blood CD4+Foxp3− T cells in WAS KO mice were not changed compared with WT mice (Figure 5A-D). After immunization, the frequency of CXCR5+ T cells within CD4+Foxp3− T cells was significantly decreased in both blood and spleen of immunized WAS KO mice compared with those in immunized WT mice (Figure 5A-D). Consistent with the Tfh cells, the percentages of memory Tfh (CXCR5+CD44hi) and Tfr cells were decreased in spleen and blood of WAS KO mice compared with those in WT mice (Figure 5E-G), but the number of Tfr cells did not change (Figure 5H).

Tfh cells in blood and spleen of immunized WAS KO and WT mice. (A) Flow cytometric analysis of B220−CD4+Foxp3−CXCR5+ cells in blood and spleen of WAS KO mice and WT mice after secondary immunization (immu). un, unimmunized control. Percentage of CXCR5+ cells within CD4+Foxp3− T cells in blood (B) and spleen (C) of WT mice and WAS KO mice. (D) Number of Tfh cells in the spleen of WT and WAS KO mice. Percentage of CXCR5+CD44hi cells within CD4+Foxp3− T cells in blood (E) and spleen (F) of WAS KO mice and WT mice. (G) Percentage and (H) number of Tfr in the spleen of WT and WAS KO mice. (I) Flow cytometric analysis of CXCR5+ICOS+ cells in spleen and blood of WT and WAS KO mice. The percentage of CXCR5+ICOS+ T cells in blood (J) and spleen (K) of WT and WAS KO mice. (L) The number of CXCR5+ICOS+ T cells in spleen of WT and WAS KO mice. (M) Flow cytometric analysis of CXCR5+BCL6+ cells in spleen of WAS KO mice and WT mice. (N) Percentage and (O) number of CXCR5+BCL6+ cells within CD4+ T cells in the spleen of WAS KO mice and WT mice.
Tfh cells in blood and spleen of immunized WAS KO and WT mice. (A) Flow cytometric analysis of B220−CD4+Foxp3−CXCR5+ cells in blood and spleen of WAS KO mice and WT mice after secondary immunization (immu). un, unimmunized control. Percentage of CXCR5+ cells within CD4+Foxp3− T cells in blood (B) and spleen (C) of WT mice and WAS KO mice. (D) Number of Tfh cells in the spleen of WT and WAS KO mice. Percentage of CXCR5+CD44hi cells within CD4+Foxp3− T cells in blood (E) and spleen (F) of WAS KO mice and WT mice. (G) Percentage and (H) number of Tfr in the spleen of WT and WAS KO mice. (I) Flow cytometric analysis of CXCR5+ICOS+ cells in spleen and blood of WT and WAS KO mice. The percentage of CXCR5+ICOS+ T cells in blood (J) and spleen (K) of WT and WAS KO mice. (L) The number of CXCR5+ICOS+ T cells in spleen of WT and WAS KO mice. (M) Flow cytometric analysis of CXCR5+BCL6+ cells in spleen of WAS KO mice and WT mice. (N) Percentage and (O) number of CXCR5+BCL6+ cells within CD4+ T cells in the spleen of WAS KO mice and WT mice.
In order to investigate whether the differentiation defect of Tfh cells in WAS KO mice is cell intrinsic, we transferred CD45.2 WT or WAS KO bone marrow cells with CD45.1 bone marrow cells at a ratio of 1:1 into irradiated CD45.1 WT recipient mice. After 8 weeks of reconstitution, the frequency of CD45.2+ Tfh cells did not differ in spleens of naive WT and WAS KO chimera mice (supplemental Figure 4A-B), but it was significantly decreased in the spleens of immunized WAS KO chimera mice compared with that of WT chimera mice (supplemental Figure 4A-B). These results suggested the differentiation defect of Tfh cells in WAS KO mice is cell intrinsic. Furthermore, we found that the frequency of CXCR5+ICOS+ T cells in both the blood and spleen of WAS KO mice was higher than in WT mice regardless of whether the animals were naive or immunized, but the number of CXCR5+ICOS+ was increased only in naive mice (Figure 5I-L). ICOS is highly expressed in activated T cells, and in order to exclude the extrinsic environment in WAS KO mice, we examined the frequency of CD45.2+CXCR5+ICOS+ T cells in chimera mice and found it was significantly decreased in the spleen of both nonimmunized and immunized WAS KO chimera mice compared with WT chimera mice (supplemental Figure 4C-D). Additionally, the frequency and number of CXCR5+BCL6+ T cells were significantly reduced in the spleen of immunized WAS KO mice compared with immunized WT mice (Figure 5M-O); therefore, the reduction of CXCR5+ICOS+ and CXCR5+BCL6+ T cells is in line with the finding of reduced Tfh cells in WAS KO mice. In order to further investigate the cellular mechanism for the reduction of Tfh cells in WAS KO mice, we examined the proliferation and apoptosis of Tfh cells in WAS patients or WAS KO mice using Ki-67 and caspase-3 antibodies. The percentage of Ki-67+ Tfh cells was decreased in Tfh cells of WAS patients (Figure 6A-B) and WAS KO mice (Figure 6C) after PHA stimulation, and the percentage of caspase-3+ Tfh cells was increased in Tfh cells of WAS patients with or without PHA stimulation (Figure 6A,D,E), but not in WAS KO mice, regardless of whether they were stimulated with PHA or not (Figure 6F-G). Additionally, a decreased frequency of Ki-67+ Tfh cells and an increased frequency of caspase-3+ Tfh cells could also be seen in WAS KO chimera mice with PHA stimulation (supplemental Figure 4E-F). These results indicate that reduced proliferation and increased apoptosis account for the reduction in Tfh cells in WAS patients and WAS KO mice and are cell autonomous.

The proliferation and apoptosis of Tfh cells in HCs and WAS patients or WT and WAS KO mice. (A) Flow cytometry analysis of caspase-3 or Ki-67 staining of Tfh cells in HCs and WAS patients stimulated without or with PHA. (B) The percentage of Ki-67+ Tfh cells in PBMCs from HCs and WAS patients stimulated with PHA. (C) The percentage of Ki-67+ Tfh cells in the spleen of WT and WAS KO mice stimulated with PHA. The percentage of caspase-3+ Tfh cells in PBMCs from HCs and WAS patients stimulated with PHA (D) or without PHA (E). The percentage of caspase-3+ Tfh cells in the spleen of WT and WAS KO mice stimulated with PHA (F) or without PHA (G).
The proliferation and apoptosis of Tfh cells in HCs and WAS patients or WT and WAS KO mice. (A) Flow cytometry analysis of caspase-3 or Ki-67 staining of Tfh cells in HCs and WAS patients stimulated without or with PHA. (B) The percentage of Ki-67+ Tfh cells in PBMCs from HCs and WAS patients stimulated with PHA. (C) The percentage of Ki-67+ Tfh cells in the spleen of WT and WAS KO mice stimulated with PHA. The percentage of caspase-3+ Tfh cells in PBMCs from HCs and WAS patients stimulated with PHA (D) or without PHA (E). The percentage of caspase-3+ Tfh cells in the spleen of WT and WAS KO mice stimulated with PHA (F) or without PHA (G).
WASp deficiency in Tfh cells causes the T-cell–intrinsic functional defect both in vitro and in vivo
In order to confirm whether the WASp deficiency in Tfh cells that causes the functional defect is T cell intrinsic, we cocultured naive B cells and Tfh cells from HCs or WAS patients or from WT or WAS KO mice pulsed with SEB. After coculture for 3 days, the immunoglobulin titer was determined by ELISA. We found the concentration of IgG was significantly decreased while that of IgM did not change in WAS patients (Figure 7A) or WAS KO mice (Figure 7B). This result suggested that WAS patients or WAS KO Tfh cells fail to promote the differentiation of B cells in vitro. In order to further determine the functional defect of WAS KO Tfh cells in vivo, we adoptively transferred WT or WAS KO CD4+ naive T cells into CD4 KO mice and then immunized the mice with NP-KLH. After 7 days, we examined the GC and memory B cells in CD4 KO recipient mice. We found the frequency and number of GC B cells were significantly reduced in CD4 KO mice transferred with WAS KO CD4+ naive T cells compared with WT CD4+ naive T cells, regardless of whether the cells were antigen specific or not (Figure 7C-F), and the reduction of GC B cells was more severe for antigen-specific B cells (Figure 7C-F). Memory B cells were also reduced in CD4 KO mice transferred with WAS KO CD4+ naive T cells (Figure 7G). Additionally, the expression levels of CD86, a B-cell activation marker, were also reduced in NP+ B cells (Figure 7H). All of these results suggest that WASp plays a critical role in the normal functioning of Tfh cells.

The in vitro and in vivo functions of WAS patient Tfh cells or WAS KO Tfh cells. ELISA assay of IgG and IgM in the culture medium from HC naive B cells cocultured with HC or WAS Tfh cells (A) or from WT naive B cells cocultured with WT or WAS KO Tfh cells pulsed with SEB at day 3 (B). Flow cytometry analysis of GC B cells (C) or NP-specific GC B cells (D) from CD4 KO mice transferred with WT or WAS KO CD4+ naive T cells at day 7. The percentage and number of GC B cells (E) or NP-specific GC B cells (F) in CD4 KO mice transferred with WT or WAS KO CD4+ naive T cells at day 7. (G) The percentage and number of memory B cells in spleen of CD4 KO mice transferred with WT or WAS KO CD4+ naive T cells at day 7. (H) The MFI of CD86 in NP-specific B cells in CD4 KO mice transferred with WT or WAS KO CD4+ naive T cells. SSC, sidescatter.
The in vitro and in vivo functions of WAS patient Tfh cells or WAS KO Tfh cells. ELISA assay of IgG and IgM in the culture medium from HC naive B cells cocultured with HC or WAS Tfh cells (A) or from WT naive B cells cocultured with WT or WAS KO Tfh cells pulsed with SEB at day 3 (B). Flow cytometry analysis of GC B cells (C) or NP-specific GC B cells (D) from CD4 KO mice transferred with WT or WAS KO CD4+ naive T cells at day 7. The percentage and number of GC B cells (E) or NP-specific GC B cells (F) in CD4 KO mice transferred with WT or WAS KO CD4+ naive T cells at day 7. (G) The percentage and number of memory B cells in spleen of CD4 KO mice transferred with WT or WAS KO CD4+ naive T cells at day 7. (H) The MFI of CD86 in NP-specific B cells in CD4 KO mice transferred with WT or WAS KO CD4+ naive T cells. SSC, sidescatter.
Discussion
The contribution of Tfh cells to the pathogenesis of WAS has not been defined. In this study, we first demonstrated that WAS patients displayed a decreased number of circulating Tfh cells along with reduced expression of the transcription factor BCL6 in CD4+ T cells. Although Tfh cells in WAS patients or WAS KO mice showed increased expression of ICOS and were capable of secreting cytokine IL-21, results in chimera mice suggested that this increase was cell extrinsic due to the environment of WAS KO mice. Furthermore, WAS KO mice showed a compromised memory response in blood and spleen, as determined by the frequency of Tfh cells after secondary immunization with NP-KLH. Finally, the in vitro coculture experiment and in vivo adoptive transfer experiment confirmed the defective function of WASp-deficient Tfh cells was T-cell intrinsic. Therefore, our present study provides evidence that the defective memory formation, maintenance, and differentiation of Tfh cells appear to be involved in the pathogenesis of immunodeficiency in WAS.
Several studies report decreased numbers of CXCR5+CD4+ T cells in the blood of patients with antibody deficiencies.12,23,24 In healthy children, the percentage of blood CXCR5+CD4+ T cells rapidly increases during the first 6 months of life and peaks at 1 to 2 years of age.25 Our study showed an obvious reduction in the proportion of blood CXCR5+CD4+ T cells in children with WAS with increasing age. Similarly, previous studies have shown reduced T- and B-cell regions in the GC as well as progressive depletion of GCs in spleens and lymph nodes from WAS patients.26,27 Although patients with autoimmunity display increased numbers of CXCR5+CD4+ T cells in the blood,28-30 in our cohort, WAS patients with a 5A score still showed a reduced frequency of CXCR5+CD4+ T cells. Notably, all patients with absent WASp expression showed a clearly reduced frequency of CXCR5+CD4+ T cells. These findings further highlighted the close relationship between WASp expression and the frequency of Tfh cells in WAS. Our results therefore indicate that WASp participates in the generation and/or maintenance of Tfh cells.
Circulating memory Tfh cells can be divided into Th1-like, Th2-like, and Th17-like subsets, with different capacities to regulate B-cell responses. Th2-like and Th17-like Tfh cells efficiently induced naive B cells to proliferate and differentiate into plasmablasts in vitro.19 Altered Tfh subsets have also been reported in autoimmune diseases, such as juvenile dermatomyositis19 and Sjögren’s syndrome.28 In our study, the skewing of blood Tfh subsets was consistently associated with autoimmune disease in WAS, and we found a higher ratio of Th2-like + Th17-like Tfh cells to Th1-like Tfh cells in patients with a clinical score of 5A and no WASp expression. In line with a previous study that showed impaired secretion of Th1 cytokines in WAS,4 we found that Th1-like Tfh cells were markedly decreased in WAS. Thus, our data suggest that the development of protective antibody responses from preexisting memory B cells is impaired in WAS.
Tfh memory is generated along with the generation of B-cell memory.31 In WAS patients, memory B cells are reduced in absolute number.32 Functionally, memory Tfh cells not only promote a primary response but also enhance a secondary response upon antigen rechallenge.31 In addition, WAS KO mice have reduced antibody production, a delayed GC reaction, and abnormal immunoglobulin class switching.33 Furthermore, WAS KO animals have a reduced secondary T-cell response to influenza, as determined by a reduction in the number of NP-specific CD8+ splenic T cells and fewer memory CD4+ T cells, compared with WT mice.34,35 We found fewer memory Tfh cells in both blood and spleen of WAS KO mice after secondary immunization with NP-KLH, which was consistent with a decrease in the number of memory Tfh cells in WAS patients. These observations suggested that WASp plays a crucial role in the memory response of Tfh cells.
The production and differentiation of Tfh cells are influenced by many factors, such as IL-21, ICOS, BCL6, and BLIMP1.13 We found no significant difference in the number of IL-21–secreting CD4+CXCR5+ cells and BLIMP1 expression between WAS patients and HCs, but consistent with the decreased number of CXCR5+CD4+ T cells in blood, the expression of BCL6 by CD4+ T cells and Tfh cells was decreased in WAS patients. BCL6 is a master regulator of the main pathway of Tfh-cell differentiation and function.10 ICOS provides a critical early signal to induce BCL6, which in turn induces CXCR5.36,37 In our study, we observed relatively lower level levels of CXCR5 and BCL6 expression but higher levels of ICOS expression in WAS patients and immunized WAS KO mice. WAS KO chimera mice showed lower levels of ICOS expression, which indicates that the increase in levels of ICOS expression in WAS KO mice was cell extrinsic due to the environment of WAS KO mice. The question remains as to how WASp regulates ICOS and BCL6 to control the differentiation and function of Tfh cells. WASp has been involved in the transcriptional events by translocating into the nucleus. The block of small ubiquitin-related modifier (SUMO)ylation converts nuclear WASp from a transcriptional coactivator into a corepressor of nuclear factor κB response genes in human TH1-differentiating cells.38 Therefore, WASp may regulate ICOS and BCL6 at the transcriptional level.
In the blood of healthy subjects, circulating CXCR5+CD4+ T cells express ICOS at very low levels, whereas in lupus patients, CD4+ICOS+ cells are present at a higher frequency,39 indicating a connection between high expression of ICOS and autoimmunity. Increased frequencies of CXCR5+PD-1hi or ICOShi cells in peripheral blood are associated with extensive autoantibody production.28,40 We also observed a high percentage of CXCR5+ICOS+ cells in the blood of WAS patients, but this did not appear to correlate with autoimmunity and WASp expression. Activated CD4+ T lymphocytes from WASp-deficient mice undergo reduced apoptosis after restimulation through the TCR.41 WAS−/− Tfh cells with higher expression of ICOS show reduced proliferation and increased apoptosis. ICOS costimulation is crucial to Tfh differentiation,12 which is also complicated, as both dendritic cells and B cells are involved.13 Further studies are required to elucidate the underlying mechanisms.
Our study sheds new light on the key role of WASp in the development and memory response of Tfh cells and provides an explanation for the pathogenesis of immunodeficiency in WAS. Furthermore, our study suggests that the absence of WASp contributes to decreased BCL6 transcription, which results in a deficient Tfh response. However, the mechanism through which WASp deficiency causes a reduced memory response has not been fully identified. Further studies are required to explain the persistence of the deficiency of the Tfh response in patients with WAS and its molecular mechanisms.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the patients, donors, and their families for their cooperation, Li-Lin Ye and Xin-Yuan Zhou for help with adoptive transfer, and Xin Li and Shi-fang Dong for help with flow cytometry.
This work was sponsored by grants from the Public Welfare Scientific Research Project of China (201402012).
Authorship
Contribution: X. Zhang and R.D. performed research, analyzed data, and wrote the paper; W.L., H.Z., Y.Z., L.Z., H.D., G.L., and L.N. provided help in performing the research; J.W., Y.A., Y.D., and Z.Z. helped obtain clinical samples; W.S. helped critically revise the manuscript; C.L. and X. Zhao conceptualized and designed the study and reviewed and revised the manuscript; and all authors approved the final manuscript as submitted and agree to be accountable for all aspects of the work.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Xiaodong Zhao, Ministry of Education Key Laboratory of Child Development and Disorders, Chongqing Key Laboratory of Child Infection and Immunity, Division of Immunology, Children’s Hospital of Chongqing Medical University, No. 136, Zhongshan 2nd Rd, Yuzhong District, Chongqing 400014, China; e-mail: zhaoxd530@aliyun.com; or Chaohong Liu, Ministry of Education Key Laboratory of Child Development and Disorders, Chongqing Key Laboratory of Child Infection and Immunity, Children’s Hospital of Chongqing Medical University, No.136, Zhongshan 2nd Rd, Yuzhong District, Chongqing 400014, China; e-mail: chaohongliu80@126.com.
