

Key Points
Patients with sickle cell disease have greater microvascular oxygen consumption rates than healthy individuals.
During sickle cell pain crisis, microvascular oxygen consumption increases further.
Abstract
Sickle cell disease is an inherited blood disorder characterized by chronic hemolytic anemia and episodic vaso-occlusive pain crises. Vaso-occlusion occurs when deoxygenated hemoglobin S polymerizes and erythrocytes sickle and adhere in the microvasculature, a process dependent on the concentration of hemoglobin S and the rate of deoxygenation, among other factors. We measured oxygen consumption in the thenar eminence during brachial artery occlusion in sickle cell patients and healthy individuals. Microvascular oxygen consumption was greater in sickle cell patients than in healthy individuals (median [interquartile range]; sickle cell: 0.91 [0.75-1.07] vs healthy: 0.75 [0.62-0.94] −ΔHbO2/min, P < .05) and was elevated further during acute pain crisis (crisis: 1.10 [0.78-1.30] vs recovered: 0.88 [0.76-1.03] −ΔHbO2/min, P < .05). Increased microvascular oxygen consumption during pain crisis could affect the local oxygen saturation of hemoglobin when oxygen delivery is limiting. Identifying the mechanisms of elevated oxygen consumption during pain crisis might lead to the development of new therapeutic interventions. This trial was registered at www.clinicaltrials.gov as #NCT01568710.
Introduction
Sickle cell disease is a blood disorder caused by homozygous or compound-heterozygous inheritance of abnormal hemoglobin-β chains that form hemoglobin S. Patients with sickle cell disease endure pain crises that may last days and occur multiple times each year.1,2 The etiology of painful crises is unknown but may involve blockage of vessels by sickled and adherent blood cells,3 followed by ischemia reperfusion injury4 and local inflammatory responses.5 Inflammation, in addition to increasing pain, can increase oxygen consumption6,7 and might have adverse effects on hemoglobin oxygenation and sickling when oxygen delivery is limiting. We hypothesized that sickle cell patients would have increased rates of oxygen consumption during acute pain crisis. We measured microvascular oxygen consumption and systemic biomarkers of inflammation in healthy African American volunteers, in patients with sickle cell disease in clinical steady state, and in patients both during pain crisis and after recovery.
Study design
Patients
The Institutional Review Board of the National Heart, Lung and Blood Institute approved clinical protocol 12-H-0101 specifically for this study. All participants provided written informed consent in accordance with the Declaration of Helsinki. See http://www.clinicaltrials.gov/ct2/show/NCT01568710 and supplemental Table 1 (available on the Blood Web site) for enrollment criteria. Pain crisis was defined as acute pain occurring in a typical distribution requiring hospital admission and parenteral analgesia. Acute crisis studies were performed within 36 hours of admission, after patients had received intravenous fluids and pain medications. Follow-up studies were performed more than 3 weeks after resolution of acute pain symptoms.
Near-infrared spectroscopy and oxygen consumption calculation
Near-infrared spectroscopy has been validated against magnetic resonance spectroscopy as a measure of local oxygen consumption in muscle.8 We used the Inspectra 650 (Hutchinson Technology, Hutchinson, MN) to record tissue hemoglobin oxygen saturation9 (StO2) and tissue hemoglobin index10 (THI; a measure of hemoglobin signal strength) every 2 seconds during a 5-minute brachial artery occlusion. Oxygen consumption (VO2) was calculated as the sum of each change in StO2 over each 2-second interval, weighted by the THI, all divided by the duration of occlusion t.
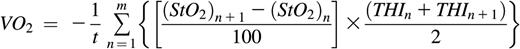
Statistics
Unpaired t tests, Mann-Whitney U tests, paired t tests, or Wilcoxon matched-pairs signed-rank tests were performed where appropriate using GraphPad Prism 6 (GraphPad Software, San Diego, CA). Data are presented as median (interquartile range).
Results and discussion
Microvascular oxygen consumption
We estimated microvascular oxygen consumption by monitoring the decline in hemoglobin oxygen saturation in the thenar eminence while preventing arterial inflow from the brachial artery. Microvascular oxygen consumption was greater among sickle cell patients in steady state (0.91 [0.75-1.07] −ΔHbO2/min) compared with healthy individuals (0.75 [0.62-0.94] −ΔHbO2/min, P < .05). Oxygen consumption was greater during acute pain crisis (1.10 [0.78-1.30] −ΔHbO2/min) compared with either steady state (0.91 [0.75-1.07] −ΔHbO2/min, P < .05) or after recovery from pain crisis using paired analysis (recovered: 0.88 [0.76-1.03] −ΔHbO2/min, P < .05), as shown in Figure 1 and Table 1. Taken together, these results suggest that oxygen consumption is chronically elevated in sickle cell patients in steady state, increases acutely during pain crisis, and then returns to a steady-state baseline after recovery from crisis.

Elevated oxygen consumption, neutrophil count, and CRP during sickle cell pain crisis. Thenar eminence microvascular oxygen consumption (A; VO2), absolute (Abs) neutrophil count (B), and CRP levels (C) were greater among sickle cell disease (SCD) patients in pain crisis than among patients in steady-state or healthy individuals. Horizontal lines indicate the median for each group. Significance levels are indicated by *P < .05, **P < .01, and ns (not significant).
Elevated oxygen consumption, neutrophil count, and CRP during sickle cell pain crisis. Thenar eminence microvascular oxygen consumption (A; VO2), absolute (Abs) neutrophil count (B), and CRP levels (C) were greater among sickle cell disease (SCD) patients in pain crisis than among patients in steady-state or healthy individuals. Horizontal lines indicate the median for each group. Significance levels are indicated by *P < .05, **P < .01, and ns (not significant).
Participant characteristics, hematologic parameters, and inflammatory markers
| Healthy volunteer | Sickle cell steady state | Sickle cell pain crisis | Recovered from pain crisis | |
|---|---|---|---|---|
| Number | 38 | 29 | 20 | 16 |
| β-globin genotype | 30 AA, 7 AS, 1 AC | 26 SS, 3 SC | 18 SS, 2 SC | 14 SS, 2 SC |
| Age (y) | 31 (26-43) | 35 (28-43) | 36 (25-45) | 36 (27-45) |
| Female (%) | 24/38 (63%) | 17/29 (59%) | 13/20 (65%) | 11/16 (69%) |
| BMI (kg/m2) | 28 (24-32) | 24 (21-26)* | 23 (21-27) | 24 (22-28) |
| Hydroxyurea use (%) | 0/38 (0%) | 20/29 (69%) | 17/20 (85%) | 14/16 (88%) |
| Hemoglobin (g/dL) | 13 (12-14) | 9.1 (8.1-9.7)* | 7.7 (7.1-8.8)† | 8.5 (7.4-9.3) |
| Fetal hemoglobin (%) | 0.0 | 9.5 (5.0-18)* | 12 (4.0-16) | 13 (2.9-16) |
| Hemoglobin S (%) | 0.0 | 76 (61-81)* | 81 (77-83) | 78 (57-83) |
| Hematocrit (%) | 38 (36-41) | 25 (22-27)* | 21 (20-24)† | 24 (21-27)‡ |
| MCV (fL) | 85 (79-90) | 93 (80-110)* | 94 (86-100) | 90 (77-100) |
| Reticulocyte (%) | 1.0 (0.8-1.5) | 8.7 (4.3-11)* | 8.1 (5.7-12) | 9.0 (6.2-14) |
| Retic count (K/uL) | 50 (36-72) | 190 (110-280)* | 170 (150-300) | 240 (140-320) |
| LDH (U/L) | 180 (160-200) | 330 (280-470)* | 480 (330-660) | 390 (350-440) |
| Platelet count (K/uL) | 240 (220-280) | 300 (230-370)* | 280 (210-380) | 310 (230-490) |
| WBC count (K/uL) | 5.4 (4.6-6.9) | 7.1 (5.9-9.5)* | 9.2 (7.6-13)† | 8.9 (7.1-14) |
| Neutrophil count (K/uL) | 3.0 (2.1-3.8) | 3.4 (2.1-5.2) | 5.7 (3.3-7.2)† | 3.6 (2.7-6.6) |
| CRP (mg/L) | 1.2 (0.4-2.9) | 3.3 (1.3-4.8)* | 12 (2.4-66)† | 6.0 (2.0-8.7)‡ |
| VO2 (−ΔHbO2/min) | 0.75 (0.62-0.94) | 0.91 (0.75-1.07)* | 1.10 (0.78-1.30)† | 0.88 (0.76-1.03)‡ |
| Healthy volunteer | Sickle cell steady state | Sickle cell pain crisis | Recovered from pain crisis | |
|---|---|---|---|---|
| Number | 38 | 29 | 20 | 16 |
| β-globin genotype | 30 AA, 7 AS, 1 AC | 26 SS, 3 SC | 18 SS, 2 SC | 14 SS, 2 SC |
| Age (y) | 31 (26-43) | 35 (28-43) | 36 (25-45) | 36 (27-45) |
| Female (%) | 24/38 (63%) | 17/29 (59%) | 13/20 (65%) | 11/16 (69%) |
| BMI (kg/m2) | 28 (24-32) | 24 (21-26)* | 23 (21-27) | 24 (22-28) |
| Hydroxyurea use (%) | 0/38 (0%) | 20/29 (69%) | 17/20 (85%) | 14/16 (88%) |
| Hemoglobin (g/dL) | 13 (12-14) | 9.1 (8.1-9.7)* | 7.7 (7.1-8.8)† | 8.5 (7.4-9.3) |
| Fetal hemoglobin (%) | 0.0 | 9.5 (5.0-18)* | 12 (4.0-16) | 13 (2.9-16) |
| Hemoglobin S (%) | 0.0 | 76 (61-81)* | 81 (77-83) | 78 (57-83) |
| Hematocrit (%) | 38 (36-41) | 25 (22-27)* | 21 (20-24)† | 24 (21-27)‡ |
| MCV (fL) | 85 (79-90) | 93 (80-110)* | 94 (86-100) | 90 (77-100) |
| Reticulocyte (%) | 1.0 (0.8-1.5) | 8.7 (4.3-11)* | 8.1 (5.7-12) | 9.0 (6.2-14) |
| Retic count (K/uL) | 50 (36-72) | 190 (110-280)* | 170 (150-300) | 240 (140-320) |
| LDH (U/L) | 180 (160-200) | 330 (280-470)* | 480 (330-660) | 390 (350-440) |
| Platelet count (K/uL) | 240 (220-280) | 300 (230-370)* | 280 (210-380) | 310 (230-490) |
| WBC count (K/uL) | 5.4 (4.6-6.9) | 7.1 (5.9-9.5)* | 9.2 (7.6-13)† | 8.9 (7.1-14) |
| Neutrophil count (K/uL) | 3.0 (2.1-3.8) | 3.4 (2.1-5.2) | 5.7 (3.3-7.2)† | 3.6 (2.7-6.6) |
| CRP (mg/L) | 1.2 (0.4-2.9) | 3.3 (1.3-4.8)* | 12 (2.4-66)† | 6.0 (2.0-8.7)‡ |
| VO2 (−ΔHbO2/min) | 0.75 (0.62-0.94) | 0.91 (0.75-1.07)* | 1.10 (0.78-1.30)† | 0.88 (0.76-1.03)‡ |
Data are presented as median (interquartile range). Statistics performed using unpaired t tests, Mann-Whitney U tests, paired t tests, or Wilcoxon matched-pairs signed rank tests, where appropriate.
BMI, body mass index; CRP, C-reactive protein; δHbO2/min, change in oxygenated hemoglobin per minute; LDH, lactate dehydrogenase; MCV, mean corpuscular volume; Retic, reticulocyte; VO2, oxygen consumption rate; WBC, white blood cell.
P < .05 for steady state compared with healthy volunteer.
P < .05 for acute crisis compared with steady state.
P < .05 for recovered from crisis compared with acute crisis.
Inflammatory biomarkers
We assessed each patient’s inflammatory state by neutrophil count and C-reactive protein (CRP) concentration. Absolute neutrophil count was elevated during acute pain crisis compared with steady state (crisis: 5.7 [3.3-7.2] vs steady state: 3.4 [2.1-5.2] K/uL, P < .01) but remained unchanged after recovery from crisis (crisis: 5.7 [3.3-7.2] to recovery: 3.6 [2.7-6.6], P = .33). CRP was acutely elevated during pain crisis compared with steady state (crisis: 12 [2.4-66] vs steady state: 3.3 [1.3-4.8] mg/L, P < .01) and decreased after resolution of crisis to 6.0 (2.0-8.7) mg/L (P < .05). Our findings of elevated inflammatory biomarkers during sickle cell pain crisis are consistent with previous studies showing elevated neutrophil count and CRP in steady state with further elevation during pain crisis, though we did not observe an elevated neutrophil count in steady state compared with healthy individuals as previously reported.13,14 The observational nature of this study does not allow us to causally link inflammation with increased oxygen consumption; however, these data emphasize the relevance of inflammation to the pathophysiology of sickle cell disease, especially during pain crisis.
Possible causes of elevated oxygen consumption
Several factors might elevate oxygen consumption in sickle cell disease and in pain crisis specifically. In steady state, patients with sickle cell disease experience elevated resting energy expenditure, requiring greater systemic oxygen consumption.15,16 This has been attributed to an increased rate of protein synthesis at sites of erythropoiesis. Although oxygen consumption was greater in steady state compared with healthy individuals (0.91 [0.75-1.07] vs 0.75 [0.62-0.94] −ΔHbO2/min, P < .05), our measurements are more likely to reflect the local density and activity of intravascular blood cells and myocytes rather than the metabolic demands of erythropoiesis at distant sites. Oxygen consumption by inflammatory cells in the blood contributes measurably to both local and systemic oxygen consumption; stimulation of phagocytes by phorbol myristate acetate elevated total body oxygen consumption by 18% in guinea pigs and was prevented by coadministration of a reduced NAD phosphate oxidase inhibitor, indicating that the respiratory burst of phagocytes was responsible for systemic changes in oxygen consumption.17 Similarly, controlled exposure to endotoxin, a potent inducer of inflammation, increased total body oxygen consumption by 39% in human volunteers.7 Our observations that sickle cell patients in pain crisis have local oxygen consumption rates that are 24% greater than steady state (P < .01) and 46% greater than healthy volunteers (P < .0001) are similar in magnitude to the changes induced by acute inflammatory stimuli such as phorbol myristate acetate and endotoxin.
In addition to the cellular activity of reduced NAD phosphate oxidase, the process of hemoglobin autoxidation may contribute importantly to microvascular oxygen consumption. Previous studies have attributed the enhanced production of reactive oxygen species in sickled erythrocytes to reactions catalyzed by hemoglobin,18 possibly augmented by increases in membrane-bound heme iron and free iron.19 In this study, we found that sickle cell patients in crisis had greater concentrations of methemoglobin in venous blood than did patients in steady state or healthy individuals (crisis: 1.70 [1.43-2.00]%, steady state: 1.40 [1.13-1.65]%, P < .01; healthy: 0.70 [0.60-0.80]%, P < .0001). This suggests an increased rate of hemoglobin autoxidation during sickle cell pain crisis, though impaired reduction of methemoglobin may also play a role.20 Mitochondrial activity could also potentially contribute to increased oxygen consumption during crisis through altered cellular respiration or generation of reactive oxygen species.21
Although oxygen consumption at the thenar eminence was elevated among patients experiencing sickle cell pain crisis, it is conceivable that oxygen consumption would be greater still at sites of pain where activated inflammatory cells would be more concentrated.22 Imaging modalities such as computed tomography and magnetic resonance imaging combined with markers of oxygen consumption might better elucidate the changes in oxygen consumption that occur at sites of pain. Nevertheless, the simplicity and safety of near-infrared spectroscopy combined with controlled brachial artery occlusion facilitated the first measurements of microvascular oxygen consumption during sickle cell pain crisis. The discovery of elevated oxygen consumption during crisis identifies a potential new target for the treatment of acute pain crisis.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Ken Chang and Stephen Yoon for their technical contributions. Research nursing care was provided by Linda Tondreau, Bisi Dada, Mijung Kim, Ick-Ho Kim, Elizabeth Witter, Wendy Holt, Mashood Esfanaji, Grace Kim, Miwha Yi, Elmer Amparo, and Stella Woo. Professional protocol management was provided by Stephanie Housel, MS, CIP and Mary Hall, CIP. The authors thank BethAnn Guthmueller, CCRP, and Hutchinson Technology for providing a near-infrared spectroscopy monitor and sensors for use in this study. Junfeng Sun provided statistical advice on the study design. The authors also acknowledge the contributions of the physicians, nurse practitioners, and nurses who provided care for the patients in this study and thank the patients for participating.
This study was supported by the Intramural Research Program of the National Institutes of Health at the National Heart, Lung and Blood Institute, the National Institute of Allergy and Infectious Diseases, and the National Institute of Biomedical Imaging and Bioengineering.
Authorship
Contribution: H.A., A.K.I., A.M.G., and G.J.K. designed the study; H.A., A.K.I., T.C.A., and J.N. developed the protocol; C.S., H.A., T.C.A., and A.K.C. scheduled, consented, and evaluated the participants; A.K.I., M.S., T.C.A., M.D.A., H.A., and A.M.G. collected data; A.K.I., L.M., C.A.R., and H.A. processed blood samples; and C.A.R. and H.A. analyzed and interpreted data and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for A.K.I. is School of Medicine & Health Sciences, The George Washington University, Washington, DC.
The current affiliation for T.C.A. is Department of Emergency Medicine, Alameda Health Systems-Highland Hospital, Oakland, CA.
The current affiliation for G.J.K. is Division of Hematology, Department of Medicine, Heart, Lung, Blood, and Vascular Medicine Institute, University of Pittsburgh, Pittsburgh, PA.
Correspondence: Hans Ackerman, Laboratory of Malaria and Vector Research, National Institute of Allergy and Infectious Diseases, 12735 Twinbrook Pkwy, Room 3E-28, Rockville, MD 20852; e-mail: hans.ackerman@nih.gov.
