

Abstract
Whole genome and exome sequencing studies have identified numerous genomic alterations in DLBCL, but these methods have limited applicability for the clinical care of lymphoma patients due to cost, specific tissue requirements, and laborious bioinformatic analysis. FoundationOne-Heme (FOH) is a novel next-generation sequencing platform designed to provide targeted assessment of the genomic landscape of hematologic malignancies, including identification of mutations within specific genes, copy number changes, and translocations. FOH can be performed on small quantities of formalin-fixed paraffin-embedded (FFPE) tissue, detect rare variants due to extensive depth of sequencing coverage, and rapidly provide results via streamlined bioinformatic interpretation. Here we report the first experience using this novel platform to evaluate the genetic landscape of DLBCL.
Genomic DNA and total RNA were isolated from FFPE tissue on a cohort of 53 cases of DLBCL, including de novo (n=30), relapsed/refractory (n=12), and large cell transformation from low-grade lymphoma (n=11). The cohort included 25 cases with combined MYC and BCL2 overexpression by IHC (criteria for positivity: >40% MYC, >70% BCL2), of which only one had a known translocation involving MYC. Adaptor ligated sequencing libraries were captured by solution hybridization using two custom bait sets targeting 374 cancer-related genes and 24 genes frequently rearranged for DNA-seq, and 258 frequently-rearranged genes for RNA-seq. All captured libraries were sequenced to high depth (Illumina HiSeq), averaging >658x for DNA and >20,000,000 total pairs for RNA, to enable the sensitive and specific detection of genomic alterations. Significant non-synonomous variants were identified as mutations from the COSMIC database, amplifications of established oncogenes, or homozygous deletions and/or clear loss-of-function mutations of known tumor suppressors.
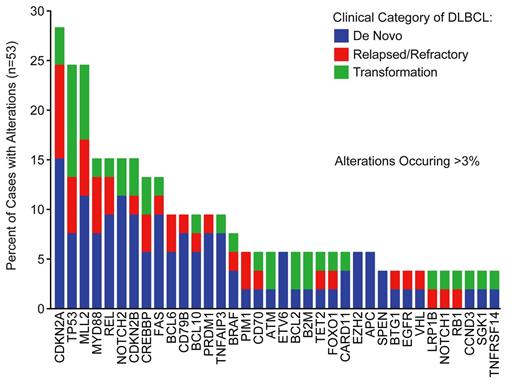
The DNA sequencing component of FOH detected translocations in BCL2, BCL6, and MYC, while the RNA sequencing component detected fusion transcripts involving BLC6 and MYC, in agreement with independent cytogenetic analysis via karyotype and FISH where available. The assay detected copy number alterations of 44 different genes, most commonly amplification of REL (15%) or loss of CDKN2A/CDKN2B (17%). The most frequent alterations of known significance are detailed in Figure 1. The most commonly altered gene was CDKN2A, exhibiting either homozygous deletion or loss of function mutation in 28% of cases. Chromatin modifying factors (e.g. MLL2, CREBBP, EZH2) represented the most frequently altered biologic category with alterations occurring in >50% of cases. Recurrent alterations in components of the Notch pathway (NOTCH1/2/4, FBXW7, SPEN), each predicted to activate the pathway, were identified in 23% of cases. Cell-of-origin was determined as per the Hans model using IHC for CD10, BCL6, and IRF4/MUM1; CD79B mutations were detected exclusively in non-GCB and EZH2 mutations were found exclusively in GCB-phenotype cases. Furthermore, IHC MYC+/BCL2+ de novo DLBCL cases (n=11) exhibited more frequent hypermutation of PIM1 (46%) compared with the 19 cases of IHC MYC-/BCL2- de novo DLBCL (11%). When comparing the various clinical categories, we found that mutations in tumor suppressors were significantly more common in relapsed/refractory than de novo DLBCL (47% vs 75%, p=0.02). Alterations in TP53 were most frequently observed in transformed lymphoma (55%).
Our results demonstrate the feasibility of using a targeted next-generation sequencing platform on FFPE clinical specimens from patients with DLBCL as a means of providing an integrated analysis of gene mutations, copy number alterations, and translocations. This streamlined approach combines multiple molecular and cytogenetic tests into a single platform and uses a small amount of tissue to perform a multifaceted assessment of genomic alterations with potential diagnostic, prognostic, and therapeutic implications. Future efforts will be directed at analyzing additional cases of DLBCL to better establish the biologic and clinical significance of the observed genetic alterations, and to prospectively incorporate this novel platform to select patients for mechanism-based targeted therapy.
Intlekofer:Foundation Medicine, Inc: Consultancy. Levine:Foundation Medicine, Inc: Consultancy. Zelenetz:Foundation Medicine, Inc: Consultancy. Palomba:Foundation Medicine, Inc: Consultancy. van den Brink:Foundation Medicine, Inc: Consultancy. Brennan:Foundation Medicine, Inc: Employment. Young:Foundation Medicine, Inc: Employment. He:Foundation Medicine, Inc: Employment. Nahas:Foundation Medicine, Inc: Employment. Yelensky:Foundation Medicine, Inc: Employment. Otto:Foundation Medicine, Inc: Employment. Lipson:Foundation Medicine, Inc: Employment. Stephens:Foundation Medicine, Inc: Employment. Miller:Foundation Medicine, Inc: Employment. Younes:Foundation Medicine, Inc: Consultancy.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal