

Abstract
Achieving high-level expansion of hematopoietic stem cells (HSCs) in vitro will have an important clinical impact in addition to enabling elucidation of their regulation. Here, we couple the ability of engineered NUP98-HOXA10hd expression to stimulate > 1000-fold net expansions of murine HSCs in 10-day cultures initiated with bulk lin−Sca-1+c-kit+ cells, with strategies to purify fetal and adult HSCs and analyze their expansion clonally. We find that NUP98-HOXA10hd stimulates comparable expansions of HSCs from both sources at ∼ 60% to 90% unit efficiency in cultures initiated with single cells. Clonally expanded HSCs consistently show balanced long-term contributions to the lymphoid and myeloid lineages without evidence of leukemogenic activity. Although effects on fetal and adult HSCs were indistinguishable, NUP98-HOXA10hd–transduced adult HSCs did not thereby gain a competitive advantage in vivo over freshly isolated fetal HSCs. Live-cell image tracking of single transduced HSCs cultured in a microfluidic device indicates that NUP98-HOXA10hd does not affect their proliferation kinetics, and flow cytometry confirmed the phenotype of normal proliferating HSCs and allowed reisolation of large numbers of expanded HSCs at a purity of 25%. These findings point to the effects of NUP98-HOXA10hd on HSCs in vitro being mediated by promoting self-renewal and set the stage for further dissection of this process.
Introduction
Hematopoietic stem cells (HSCs) are rare cells within the hematopoietic hierarchy responsible for the permanent establishment of hematopoiesis.1 The ability of HSCs to self-renew is essential for their expansion throughout hematopoietic development, homeostasis, after bone marrow (BM) transplantation, and/or in response to different physiologic stresses.2-7 Elucidating the critical elements underlying this process thus has great clinical relevance.8,9 Numerous molecular elements have been shown to be essential to the self-renewal machinery of HSCs, including intracellular signaling molecules, cell cycle regulators, chromatin modifiers, and transcription factors.10,11 In addition, various growth factor receptors have been identified as mediators of environmental cues that can modulate the self-renewal process.12-17 Forced manipulation of intrinsic cellular elements also has been used successfully as an alternative and potentially complementary track to amplify HSC populations ex vivo.18-22 The latter approach includes the engineered expression of HOXB418,23,24 and various potent NUP98-HOX fusion constructs.25,26 These studies also showed the homeobox domain, but not its flanking sequences, to be an essential element in the NUP98-HOXA10hd fusion construct.21 Greatly increasing the potential clinical implications of this work, in spite of its ability to rapidly stimulate very large expansions of adult bm HSC numbers, no evidence of perturbed HSC function or regulation in vivo has been identified.
There remain many outstanding questions pertaining to the cellular targets and biologic mechanisms that lead to the production of expanded HSC numbers from NUP98-HOXA10hd–treated BM cells. To address these issues, we designed a series of experiments in which highly purified suspensions of fetal and adult bm HSCs were used as targets in both bulk and single-cell expansion culture protocols. The results demonstrate an ability of NUP98-HOXA10hd to reproducibly stimulate multilog expansions from individual HSCs, under multiple growth factor stimulatory conditions and without effects on the growth kinetics or phenotype associated with proliferating normal HSCs. These findings support an ability of NUP98-HOXA10hd to directly potentiate the self-renewal machinery operative in HSCs throughout ontogeny and introduce the reality of large-scale recovery of functionally normal HSCs at high purity.
Methods
Mice
Mice were bred and maintained at the British Columbia Cancer Agency Research Center animal facility according to the guidelines of the Canadian Council on Animal Care. Transplant donors were C57Bl/6J (B6) mice that express CD45.2 or C57Bl/6Ly-Pep3b (Pep3b) mice that express CD45.1. Recipients of B6 cells were either C57Bl/6-W41/W41 (W41) mice that express CD45.2 or Pep3b mice; recipients of Pep3b cells were B6 mice. B6 heterozygous mice expressing both CD45.1 and CD45.2 were used as recipients in certain competition assays.
Flow cytometry
Populations of cells with a lin−Sca1+kit+ (LSK), lin−Sca-1+CD43+(c-kit+)Mac1dim, CD45midlin−Rhodamine-123(Rho)− side population (SP) or CD45+EPCR+CD48−CD150+ (E-SLAM) phenotype were isolated using an FACSVantage, FACSDiVa, or AriaII cell sorter (BD Biosciences) as described previously.27-29 For transduced cells, anti-CD11b (Mac1) was left out of the lin cocktail. Data were analyzed using FlowJo Version 8.86 software (TreeStar).
Viral transduction and cell culture
GP+E86 cells producing high-titer helper virus-free MSCV-IRES-GFP (GFP) and MSCV-NUP98-HOXA10hd-IRES-GFP (NUP98-HOXA10hd) virus26,30 were seeded into the wells of round-bottomed tissue culture-treated 96-well plates at 4 × 104 irradiated (40 Gy of X-rays) cells per well, and then single or aliquots of 20 to 50 highly purified HSCs were added. Cells were cocultivated for 48 hours in 100 μL of DMEM supplemented with 15% FBS, 5 μg/mL protamine sulfate (Sigma-Aldrich), and either 10 ng/mL human IL-6, 6 ng/mL murine IL-3, and 100 ng/mL murine stem cell factor (SCF) for adult bm cell transductions or 50 ng/mL murine SCF for fetal liver cell transductions.31 Media, serum, and growth factors were all from Stem Cell Technologies. The cells from each well were then recovered, transferred to nontissue culture 96-well plates, and further cultured in 200 μL of the same medium without protamine sulfate until becoming confluent (∼ day 7 for clonal or day 4 or 5 for bulk cultures). Thereafter, on average every 2 to 3 days, cells were expanded in increasing volumes up to 2 mL of media by day 14 for clonal cultures and day 10 for bulk cultures, except for the factorial design experiments where a different experimental design was used (see “Factorial design experiments”).
Microfluidic cultures
A microfluidic cell culture array containing 1600 chambers (4.1 nl each) and an integrated iso-osmotic reservoir was fabricated as reported previously.32 Two days before loading cells into the array, the iso-osmotic bath was filled with DMEM, and the microfluidic array was primed with culture medium (DMEM supplemented with 15% FBS, 10 ng/mL IL-6, 6 ng/mL IL-3, and 100 ng/mL SCF). The environmental chamber (Chamlide; Live Cell Instrument) was set to 37°C and 5% CO2 and humidity levels were maintained close to saturation. Just before loading the cells, the bath content was replaced and the medium inside the device was exchanged. Two days after the end of infection, cells were harvested, centrifuged at 250g for 5 minutes, and resuspended in 5 to 10 μL. Using a Teflon tube connected to a stainless steel pin, control GFP-transduced cells were first introduced into the device at a rate of 1 μL/minute until the first cells reached the end of the array. The pump was then stopped to allow these cells to settle down by gravity to the bottom of the chambers. This process was repeated until the entire inoculum volume was loaded. Medium was then pumped through the array to flush out any unsettled cells. A picture set of the entire array was taken to identify wells inoculated with single control GFP-transduced cells. NUP98-HOXA10hd–transduced cells were then introduced into the device in a similar manner, and a second picture set was taken to identify wells inoculated with single cells (either control GFP- or NUP98-HOXA10hd–transduced cells). Brightfield pictures of wells initially identified as containing single cells at the start were then acquired every 5 minutes using an Axiovert 200 microscope (Carl Zeiss) outfitted with an automated stage (ProScan II; Prior Scientific), a 20× objective and an Orca ER charge-coupled device camera (Hamamatsu). A media bottle was connected to the device, and medium was exchanged automatically every 3 hours for 72 hours. At the end of the experiment, the clones were imaged using brightfield and green fluorescence to identify transduced cells.
Transplantation and analysis of transplanted mice
HSCs numbers were quantified as described previously30 by performing limiting dilution competitive repopulating unit (CRU) assays either in sublethally irradiated W41 mice (360 cGy of X-rays) or in lethally irradiated B6 or Pep3b mice (810 cGy of X-rays, given a life-sparing injection of 105 normal B6 or Pep3b BM cells). For the competitive transplant experiments, mice were injected with cell numbers from each source that were estimated to contain 30 HSCs based on previously documented HSC frequencies in fresh fetal liver,33 fresh adult bm,3 and day 10 NUP98-HOXA10hd–transduced adult BM cells (supplemental Tables 5 and 6, available on the Blood Web site; see the Supplemental Materials link at the top of the online article). Mice were bled 2, 4, and 5-6 months after transplantation and analyzed for the presence of GFP+ T- and B-lymphoid and granulocyte/monocyte white blood cells after ammonium chloride lysis of the erythrocytes, as described previously.21 Recipients with ≥ 1% donor-derived (GFP+) lympho-myeloid cells at 20 weeks after transplant, 24 weeks after transplantation, or both were considered to be repopulated by HSCs.
Factorial design experiments
E-SLAM cells were transduced, and after 7 days in standard bulk expansion cultures, lin−Sca-1+CD43+(c-kit+)Mac1dim cells were isolated and subdivided into 20 equal aliquots that were then further cultured for 7 additional days in all combinations of 4 growth factors. In total, 20 conditions were analyzed in 24 full factorial combinations plus 4 center points. The high-level growth factor concentrations were 6 ng/mL for IL-3, 10 ng/mL for IL-6, 100 ng/mL for SCF, and 100 ng/mL for human thrombopoietin (TPO; Genentech). The center point levels were 1:10 dilutions of the high concentrations, and the low levels were 0 ng/mL of each growth factor. The full array of conditions tested is presented in supplemental Table 7. Two types of endpoints were analyzed: (1) the number of lin−Sca-1+CD43+(c-kit+)Mac1dim cells present at the end of the cultures, and (2) the fraction of GFP+ cells found in the circulating white blood cells of transplanted mice after 6 weeks or 4 months. When no reconstitution was achieved, the fraction of reconstitution was defined as 0%. For full factorial modeling, the growth factor concentrations were converted into scaled estimates via the following equation, approximating the low level absence of factors as 1:100 dilutions of the high growth factor levels.
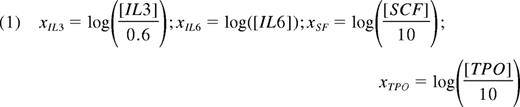
The full factorial model analyzed using JMP 8.0.2 software was then reduced to include only main and interaction effects that were significant (P < .05) for at least 1 of the 3 responses measured. After model reduction, the prediction equation obtained was of the following form:
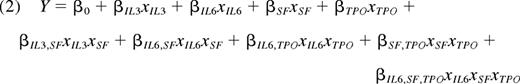
where Y is the modeled response; βi values represent main factor effects (eg, βIL3 is the main effect of IL-3); βij values represent second order interaction effects (eg, the βIL6,SF is the interaction effect of IL-6 and SCF); and βIL6,SF,TPO represents the third order interaction of IL-6, SCF, and TPO. Each response was modeled separately and the model intercept β0 represents the predicted response Y obtained using center point levels of all 4 growth factors in combination.
Viral integration analysis
Southern blot analyses were performed as described previously21 on genomic DNA isolated using DNAzol reagent (Invitrogen) and digested with EcoR1 that cleaves the integrated provirus at a single site. Digested DNA was then separated in a 0.8% agarose gel by electrophoresis and transferred to ζ-probe membranes (Bio-Rad Laboratories). Membranes were probed with a [32P]dCTP-labeled GFP sequence.
Results
Efficient high-level expansion of HSCs in single-cell cultures of NUP98-HOXA10hd–transduced CD45+lin−Rho−SP or E-SLAM cells
Single CD45+lin−Rh−SP or E-SLAM cells containing HSCs at purities of 20% to 50%29,34 were isolated by FACS, individually transduced with a NUP98-HOXA10hd or GFP control retroviral vector, and then further cultured with IL-3, IL-6, and SCF for a total of 14 days before measuring the total and CRU content of each individual culture as shown schematically in Figure 1A. Large and equivalent (∼106-fold) clonal expansions of total nucleated cells were obtained from both control and NUP98-HOXA10hd–transduced input cells (Figure 1B). However, whereas none of the control GFP-transduced clones (n = 16) contained detectable HSCs, based on the largest fractions tested (1/5 of each clonal culture), HSCs were present at high frequencies in 25 of 28 (89%) clones initiated from NUP98-HOXA10hd–transduced CD45+lin−Rho−SP cells and in 15 of 17 (88%) clones initiated from NUP98-HOXA10hd–transduced E-SLAM cells. For many of these, 1/1000 or 1/2000 of the cells in each clone were not yet at limiting dilution and showed robust long-term lympho-myeloid reconstitution of all recipients injected (Figure 1C). The average minimal HSC expansion in vitro achieved for all HSC clones tested was > 1000-fold, as shown in Figure 1E and further summarized in Table 1. The effect of NUP98-HOXA10hd overexpression on each HSC clone tested is presented in supplemental Tables 1 to 4. Analysis of vector integration sites in Southern blots of bm DNA from representative recipients confirmed the clonal origin of the expanded HSCs in each well (Figure 1D). Moreover, no difference in magnitude of HSC expansion or reconstitution level achieved for similar transplant dose was observed as a function of measured proviral copy number (eg, of clone 16 vs clone 17). From this we conclude that sufficient levels of NUP98-HOXA10hd expression were achieved on single integration and that expansion was not overtly affected by increased levels of expression.

NUP98-HOXA10hd promotes multilog expansion in vitro of individually purified adult bm HSCs. (A) Schematic of the experimental design followed to directly investigate the capacity of NUP98-HOXA10hd to enhance clonal HSC expansion in vitro. TNCs indicates total nucleated cells. (B) Average number of viable nucleated GFP- (gray) or NUP98-HOXA10hd–transduced (black) cells at the end of 14-day culture. Values shown are the means ± SD of 16 GFP- or 40 NUP98-HOXA10hd–transduced cell counts. (C) Proportion of peripheral blood leukocytes produced by a fraction (indicated as a transplantation dose) of NUP98-HOXA10hd–transduced and expanded CD45+lin−Rho−SP (top panel) and E-SLAM (bottom panel) clones transplanted into irradiated recipients 20 weeks previously. Each triangle represents an individual recipient/mouse. Solid triangles indicate recipients of the smallest fraction(s) tested of each clone, positive for the presence of transplanted cells. Open triangles indicate recipients of the highest fraction(s) tested of each clone, negative for the presence of transplanted cells. Symbol (†) indicates a dead mouse. (D) DNA extracted from NUP98-HOXA10hd–transduced bone marrow cells of representative long-term reconstituted (24-week) recipients (as shown in C top panel) was analyzed for proviral integrants by Southern blotting. Arrows indicate common integration pattern(s) among recipients of the cells from the same HSC-expanded clone (NUP98-HOXA10hd–transduced clones 16, 17, 18, 20, and 21). Transplantation dose is expressed in fraction of the culture. (E) Light gray bar indicates the maximal number of HSCs generated by day 14 in cultures of GFP-transduced cells, estimated by complete lack of reconstitution in recipients of 1/5 of 16 such cultures. Dark gray bar indicates the number of HSCs generated by day 14 in cultures of NUP98-HOXA10hd–transduced cells, as determined by limiting dilution CRU assay. Value shown is the means ± SEM of results from 40 NUP98-HOXA10hd–transduced clones tested. The presented result is the minimal number of HSCs estimated because the limiting dilution was not reached for the majority of NUP98-HOXA10hd–transduced HSC clones tested.
NUP98-HOXA10hd promotes multilog expansion in vitro of individually purified adult bm HSCs. (A) Schematic of the experimental design followed to directly investigate the capacity of NUP98-HOXA10hd to enhance clonal HSC expansion in vitro. TNCs indicates total nucleated cells. (B) Average number of viable nucleated GFP- (gray) or NUP98-HOXA10hd–transduced (black) cells at the end of 14-day culture. Values shown are the means ± SD of 16 GFP- or 40 NUP98-HOXA10hd–transduced cell counts. (C) Proportion of peripheral blood leukocytes produced by a fraction (indicated as a transplantation dose) of NUP98-HOXA10hd–transduced and expanded CD45+lin−Rho−SP (top panel) and E-SLAM (bottom panel) clones transplanted into irradiated recipients 20 weeks previously. Each triangle represents an individual recipient/mouse. Solid triangles indicate recipients of the smallest fraction(s) tested of each clone, positive for the presence of transplanted cells. Open triangles indicate recipients of the highest fraction(s) tested of each clone, negative for the presence of transplanted cells. Symbol (†) indicates a dead mouse. (D) DNA extracted from NUP98-HOXA10hd–transduced bone marrow cells of representative long-term reconstituted (24-week) recipients (as shown in C top panel) was analyzed for proviral integrants by Southern blotting. Arrows indicate common integration pattern(s) among recipients of the cells from the same HSC-expanded clone (NUP98-HOXA10hd–transduced clones 16, 17, 18, 20, and 21). Transplantation dose is expressed in fraction of the culture. (E) Light gray bar indicates the maximal number of HSCs generated by day 14 in cultures of GFP-transduced cells, estimated by complete lack of reconstitution in recipients of 1/5 of 16 such cultures. Dark gray bar indicates the number of HSCs generated by day 14 in cultures of NUP98-HOXA10hd–transduced cells, as determined by limiting dilution CRU assay. Value shown is the means ± SEM of results from 40 NUP98-HOXA10hd–transduced clones tested. The presented result is the minimal number of HSCs estimated because the limiting dilution was not reached for the majority of NUP98-HOXA10hd–transduced HSC clones tested.
Summary of all highly purified HSC clones tested and average HSC expansion in vitro achieved in response to forced expression of NUP98-HOXA10hd
| Phenotype tested | CD45+lin−Rho−SP | E-SLAM |
|---|---|---|
| No. of NUP98-HOXA10hd HSC clones tested | 28 | 17 |
| No. of expanded HSC clones | 25 | 15 |
| Minimum cell dose (fraction of culture) transplanted | 1/500 or 1/1000, or 1/2000 or 1/5000 | 1/500 or 1/1000 |
| Average minimum HSC expansion in vitro estimated (LD not reached) | > 806-fold | > 942-fold |
| Range based on ± SEM | 381 ± 1730 | 425 ± 2110 |
| Average HSC expansion in vitro documented (LD reached) | 2520-fold | |
| Range based on ± SEM | 1140 ± 5720 |
| Phenotype tested | CD45+lin−Rho−SP | E-SLAM |
|---|---|---|
| No. of NUP98-HOXA10hd HSC clones tested | 28 | 17 |
| No. of expanded HSC clones | 25 | 15 |
| Minimum cell dose (fraction of culture) transplanted | 1/500 or 1/1000, or 1/2000 or 1/5000 | 1/500 or 1/1000 |
| Average minimum HSC expansion in vitro estimated (LD not reached) | > 806-fold | > 942-fold |
| Range based on ± SEM | 381 ± 1730 | 425 ± 2110 |
| Average HSC expansion in vitro documented (LD reached) | 2520-fold | |
| Range based on ± SEM | 1140 ± 5720 |
LD indicates limiting dilution.
In vitro growth kinetics of NUP98-HOXA10hd–transduced HSCs is not perturbed
The NUP98-HOXA10hd–induced expansion of highly purified HSCs in vitro could be mediated by an increased frequency of symmetric self-renewal divisions, a decrease in HSC cell cycle transit times, an increase in HSC survival, or a combination of these effects. The generation of similar outputs of total nucleated cells by control and NUP98-HOXA10hd–transduced HSC clones suggested that the cell cycle transit times and survival (> 90% of transduced clones survived) of the NUP98-HOXA10hd–transduced cells throughout the culture period were not markedly altered. To investigate this question with greater resolution, we imaged the initial division kinetics of the transduced cells in a microfluidic array system32 (Figure 2A). The results show that highly purified (E-SLAM) HSCs transduced either with NUP98-HOXA10hd or a GFP control vector divide with the same (on average, 15-hour) cell cycle transit time (Figure 2B).

In vitro cell division kinetics of highly purified HSCs transduced either with control or NUP98-HOA10hd vector. (A) Time-lapse imaging of NUP98-HOXA10hd–transduced HSC divisions in a chamber. (B) E-SLAM cells were transduced with NUP98-HOXA10hd or GFP control retroviral vector and cultured under conditions given in Figure 3A. Two days after retroviral infection in the conventional culture, single transduced cells (42 of NUP98-HOXA10hd+ and 78 of GFP+) were deposited into individual chambers of microfluidic cell culture system. Time-lapse imaging and automated image analysis were used to score first, second, and third division of each single cell over 72 hours. Thus, first division refers not to actual first division in the culture but to first division that occurred in the microfluidic device, from which subsequent divisions were tracked and cell cycle kinetics measured. Each symbol shows the proportion of single viable cells initially deposited into individual chambers of microfluidic cell culture system that had divided by the time point shown.
In vitro cell division kinetics of highly purified HSCs transduced either with control or NUP98-HOA10hd vector. (A) Time-lapse imaging of NUP98-HOXA10hd–transduced HSC divisions in a chamber. (B) E-SLAM cells were transduced with NUP98-HOXA10hd or GFP control retroviral vector and cultured under conditions given in Figure 3A. Two days after retroviral infection in the conventional culture, single transduced cells (42 of NUP98-HOXA10hd+ and 78 of GFP+) were deposited into individual chambers of microfluidic cell culture system. Time-lapse imaging and automated image analysis were used to score first, second, and third division of each single cell over 72 hours. Thus, first division refers not to actual first division in the culture but to first division that occurred in the microfluidic device, from which subsequent divisions were tracked and cell cycle kinetics measured. Each symbol shows the proportion of single viable cells initially deposited into individual chambers of microfluidic cell culture system that had divided by the time point shown.
Proliferation phenotype of NUP98HOXA10hd–expanded HSCs allows their isolation at high purity
We next asked whether the NUP98-HOXA10hd–expanded HSCs also would show a phenotype expected of HSCs that could allow their isolation in large numbers at high purity. To address this question, we initiated cultures with 20 to 30 adult BM E-SLAM cells (estimated to contain ∼ 10-15 HSCs), transduced them with NUP98-HOXA10hd or GFP, and then cultured them for 10 days in total. The cells were then harvested, and the HSC content was measured by CRU assays of either total or various FACS-purified subpopulations (Figure 3A).
![Figure 3. CD150+CD48− subset of day 10 NUP98-HOXA10hd–transduced and expanded population contains less than half of the total HSCs generated in vitro. (A) Schematic of the experimental design where after 10-day NUP98-HOXA10hd–induced HSC expansion in vitro, bulk or purified cells were isolated based on the phenotype indicated in panel B or Figure 4A and assayed for HSC content by LDA. Because cultures were depleted of HSCs by day 10, cultures containing GFP-transduced control cells were only used for immunophenotyping and comparison and not for further transplantation assays. (B) Representative profiles of viable, Lin− NUP98-HOXA10hd–, and GFP-transduced day 10 cells, assessed for CD150 and CD48 expression. (C) Empty bars indicate the average number (expressed as the means ± SD) of bulk or phenotypically defined NUP98-HOXA10hd–transduced cells generated at the end of 10-day culture. TNCs indicates total nucleated cells. Filled bars indicate the number of HSCs (expressed as the means ± SEM) generated by day 10 in cultures of NUP98-HOXA10hd–transduced cells and the HSC content (± SEM) of phenotypically defined subsets [CD150+CD48−; CD150+CD48+; CD150−(CD48+CD48−)], as determined by limiting dilution CRU assay. (D) The distribution of the total NUP98-HOXA10hd–transduced HSCs generated in 10-day culture according to indicated phenotypes [CD150+CD48−; CD150+CD48+; CD150−(CD48+CD48−)]. Each subset was isolated by FACS and assayed for the HSC content by limiting dilution. Black area represents the HSC content (< 10% of the total) undetected by indicated phenotype(s).](/view-large/figure/7193897/zh89991179880003.jpeg)
CD150+CD48− subset of day 10 NUP98-HOXA10hd–transduced and expanded population contains less than half of the total HSCs generated in vitro. (A) Schematic of the experimental design where after 10-day NUP98-HOXA10hd–induced HSC expansion in vitro, bulk or purified cells were isolated based on the phenotype indicated in panel B or Figure 4A and assayed for HSC content by LDA. Because cultures were depleted of HSCs by day 10, cultures containing GFP-transduced control cells were only used for immunophenotyping and comparison and not for further transplantation assays. (B) Representative profiles of viable, Lin−NUP98-HOXA10hd–, and GFP-transduced day 10 cells, assessed for CD150 and CD48 expression. (C) Empty bars indicate the average number (expressed as the means ± SD) of bulk or phenotypically defined NUP98-HOXA10hd–transduced cells generated at the end of 10-day culture. TNCs indicates total nucleated cells. Filled bars indicate the number of HSCs (expressed as the means ± SEM) generated by day 10 in cultures of NUP98-HOXA10hd–transduced cells and the HSC content (± SEM) of phenotypically defined subsets [CD150+CD48−; CD150+CD48+; CD150−(CD48+CD48−)], as determined by limiting dilution CRU assay. (D) The distribution of the total NUP98-HOXA10hd–transduced HSCs generated in 10-day culture according to indicated phenotypes [CD150+CD48−; CD150+CD48+; CD150−(CD48+CD48−)]. Each subset was isolated by FACS and assayed for the HSC content by limiting dilution. Black area represents the HSC content (< 10% of the total) undetected by indicated phenotype(s).
CD150+CD48− subset of day 10 NUP98-HOXA10hd–transduced and expanded population contains less than half of the total HSCs generated in vitro. (A) Schematic of the experimental design where after 10-day NUP98-HOXA10hd–induced HSC expansion in vitro, bulk or purified cells were isolated based on the phenotype indicated in panel B or Figure 4A and assayed for HSC content by LDA. Because cultures were depleted of HSCs by day 10, cultures containing GFP-transduced control cells were only used for immunophenotyping and comparison and not for further transplantation assays. (B) Representative profiles of viable, Lin−NUP98-HOXA10hd–, and GFP-transduced day 10 cells, assessed for CD150 and CD48 expression. (C) Empty bars indicate the average number (expressed as the means ± SD) of bulk or phenotypically defined NUP98-HOXA10hd–transduced cells generated at the end of 10-day culture. TNCs indicates total nucleated cells. Filled bars indicate the number of HSCs (expressed as the means ± SEM) generated by day 10 in cultures of NUP98-HOXA10hd–transduced cells and the HSC content (± SEM) of phenotypically defined subsets [CD150+CD48−; CD150+CD48+; CD150−(CD48+CD48−)], as determined by limiting dilution CRU assay. (D) The distribution of the total NUP98-HOXA10hd–transduced HSCs generated in 10-day culture according to indicated phenotypes [CD150+CD48−; CD150+CD48+; CD150−(CD48+CD48−)]. Each subset was isolated by FACS and assayed for the HSC content by limiting dilution. Black area represents the HSC content (< 10% of the total) undetected by indicated phenotype(s).
Initial immunophenotyping confirmed a greatly reduced content of Lin+ cells in the cultures of NUP98-HOXA10hd–transduced cells20-22 indicative of the differentiation block they experience (Figures 3B and 4A) and expected HSC expansions during the 10-day culture period of ∼ 2000-fold were confirmed by limiting dilution assay (LDA) transplantation studies (Figures 3C and 4B; supplemental Tables 5-6).
Phenotypic analysis of the Lin− subset in the cells harvested from the 10-day cultures revealed up to 10% of the NUP98-HOXA10hd–transduced cells were CD150+CD48−, whereas this subset was undetectable in the Lin− cells derived from control vector-transduced cells (Figure 3B). However, only 37% of the recovered HSCs in the cultures of NUP98-HOXA10hd–transduced cells were present in the Lin−CD150+CD48− subset, and the remaining HSCs were equally distributed between the corresponding CD150+CD48+ and CD150− subsets (Figure 3C-D; supplemental Table 5).
To identify a phenotype that might give a greater enrichment and recovery of in vitro–expanded NUP98-HOXA10hd–transduced HSCs, we examined a different phenotype previously described as suitable for purifying HSCs from embryonic day 14.5 fetal livers.27,33,35,36 FACS analysis showed cells with this lin−Sca-1+CD43+(c-kit+)Mac1dim phenotype represented ∼ 4% of the Lin− population in the cultures of NUP98-HOXA10hd–transduced cells (Figure 4A). In contrast, they were barely detectable in the control cell cultures. Isolation and LDA transplantation assays of this fraction showed that it contained HSCs at a frequency of 1 in 4 and contained ∼ 87% of the total in vitro–expanded HSCs (Figure 4B-C; supplemental Table 6). High level enrichment of HSCs in the lin−Sca-1+CD43+(c-kit+)Mac1dim fraction was further confirmed by transplanting 5 cells with this phenotype (1.25 HSCs) into multiple irradiated recipients. All 10 long-term chimeras obtained in the 12 mice injected with such transplants showed a balanced lympho-myeloid contribution to the circulating peripheral blood cells up to 6 months after transplant (Figure 4D). Similar enrichments were achieved when NUP98-HOXA10hd–expanded cells were repurified using a combination of Lin/Sca-1/CD43 with SLAM markers. Three of 4 mice transplanted with 5 expanded cells of a Lin−Sca-1+CD43+CD150+CD48− phenotype showed long-term lympho-myeloid reconstitution (average level of reconstitution 13.2 ± 8%), and the calculated frequency of NUP98-HOXA10hd–transduced HSCs in this subset was 1 in 4 (1 in 2 to 1 in 7).
![Figure 4. Proliferation phenotype of NUP98HOXA10hd–expanded HSCs. (A) Representative profile of viable, Lin− NUP98-HOXA10hd– and GFP-transduced day 10 cells assessed for Sca-1 and CD43 expression and further analyzed for the level of surface Mac1 protein expression. Viable, Lin− transduced cells expressing both Sca-1 and CD43 also were confirmed to express the high levels of c-kit protein.31 (B) Empty bars indicate the average number (expressed as the means ± SD) of bulk or phenotypically defined NUP98-HOXA10hd–transduced cells at the end of 10-day culture. TNCs indicates total nucleated cells. Filled bars indicate the total number of HSCs generated by day 10 in cultures of NUP98-HOXA10hd–transduced cells and the HSC content of phenotypically defined subsets (Lin−Sca-1+c-kit+CD43+Mac1dim; Lin−Sca-1+c-kit+CD43+Mac1−; Lin−Sca-1+c-kit+CD43−Mac1bright), as determined by limiting dilution CRU assay. Values shown are the means ± SEM. (C) Distribution of the total NUP98-HOXA10hd–transduced HSCs generated in 10-day culture according to indicated phenotypes: Lin−Sca-1+c-kit+CD43+Mac1dim; Lin−Sca-1+c-kit+CD43+Mac1−; Lin−Sca-1+c-kit+CD43−Mac1bright. Each subset was isolated by FACS and assayed for the HSC content by limiting dilution. Black area represents the HSCs (< 10% of the total) undetected by indicated phenotype(s). (D) On the left, proportion of peripheral blood leukocytes produced more than 20 weeks after transplant by 5 Lin−Sca-1+c-kit+CD43+Mac1dim NUP98-HOXA10hd–transduced and expanded cells. Each triangle represents an individual recipient/mouse. On the right, bars indicate the mean percentage of lineage (Ly6G and Mac1 [black]; B220 [dark gray]; and CD4 and CD8 [light gray]) contribution to white blood cells (WBCs) of mice reconstituted with the limited number (5 cells/mouse) of NUP98-HOXA10hd–transduced and expanded cells, repurified for Lin−Sca-1+c-kit+CD43+Mac1dim phenotype (shown on the left). Error bars indicate the SD of the mean.](/view-large/figure/7193901/zh89991179880004.jpeg)
Proliferation phenotype of NUP98HOXA10hd–expanded HSCs. (A) Representative profile of viable, Lin−NUP98-HOXA10hd– and GFP-transduced day 10 cells assessed for Sca-1 and CD43 expression and further analyzed for the level of surface Mac1 protein expression. Viable, Lin− transduced cells expressing both Sca-1 and CD43 also were confirmed to express the high levels of c-kit protein.31 (B) Empty bars indicate the average number (expressed as the means ± SD) of bulk or phenotypically defined NUP98-HOXA10hd–transduced cells at the end of 10-day culture. TNCs indicates total nucleated cells. Filled bars indicate the total number of HSCs generated by day 10 in cultures of NUP98-HOXA10hd–transduced cells and the HSC content of phenotypically defined subsets (Lin−Sca-1+c-kit+CD43+Mac1dim; Lin−Sca-1+c-kit+CD43+Mac1−; Lin−Sca-1+c-kit+CD43−Mac1bright), as determined by limiting dilution CRU assay. Values shown are the means ± SEM. (C) Distribution of the total NUP98-HOXA10hd–transduced HSCs generated in 10-day culture according to indicated phenotypes: Lin−Sca-1+c-kit+CD43+Mac1dim; Lin−Sca-1+c-kit+CD43+Mac1−; Lin−Sca-1+c-kit+CD43−Mac1bright. Each subset was isolated by FACS and assayed for the HSC content by limiting dilution. Black area represents the HSCs (< 10% of the total) undetected by indicated phenotype(s). (D) On the left, proportion of peripheral blood leukocytes produced more than 20 weeks after transplant by 5 Lin−Sca-1+c-kit+CD43+Mac1dimNUP98-HOXA10hd–transduced and expanded cells. Each triangle represents an individual recipient/mouse. On the right, bars indicate the mean percentage of lineage (Ly6G and Mac1 [black]; B220 [dark gray]; and CD4 and CD8 [light gray]) contribution to white blood cells (WBCs) of mice reconstituted with the limited number (5 cells/mouse) of NUP98-HOXA10hd–transduced and expanded cells, repurified for Lin−Sca-1+c-kit+CD43+Mac1dim phenotype (shown on the left). Error bars indicate the SD of the mean.
Proliferation phenotype of NUP98HOXA10hd–expanded HSCs. (A) Representative profile of viable, Lin−NUP98-HOXA10hd– and GFP-transduced day 10 cells assessed for Sca-1 and CD43 expression and further analyzed for the level of surface Mac1 protein expression. Viable, Lin− transduced cells expressing both Sca-1 and CD43 also were confirmed to express the high levels of c-kit protein.31 (B) Empty bars indicate the average number (expressed as the means ± SD) of bulk or phenotypically defined NUP98-HOXA10hd–transduced cells at the end of 10-day culture. TNCs indicates total nucleated cells. Filled bars indicate the total number of HSCs generated by day 10 in cultures of NUP98-HOXA10hd–transduced cells and the HSC content of phenotypically defined subsets (Lin−Sca-1+c-kit+CD43+Mac1dim; Lin−Sca-1+c-kit+CD43+Mac1−; Lin−Sca-1+c-kit+CD43−Mac1bright), as determined by limiting dilution CRU assay. Values shown are the means ± SEM. (C) Distribution of the total NUP98-HOXA10hd–transduced HSCs generated in 10-day culture according to indicated phenotypes: Lin−Sca-1+c-kit+CD43+Mac1dim; Lin−Sca-1+c-kit+CD43+Mac1−; Lin−Sca-1+c-kit+CD43−Mac1bright. Each subset was isolated by FACS and assayed for the HSC content by limiting dilution. Black area represents the HSCs (< 10% of the total) undetected by indicated phenotype(s). (D) On the left, proportion of peripheral blood leukocytes produced more than 20 weeks after transplant by 5 Lin−Sca-1+c-kit+CD43+Mac1dimNUP98-HOXA10hd–transduced and expanded cells. Each triangle represents an individual recipient/mouse. On the right, bars indicate the mean percentage of lineage (Ly6G and Mac1 [black]; B220 [dark gray]; and CD4 and CD8 [light gray]) contribution to white blood cells (WBCs) of mice reconstituted with the limited number (5 cells/mouse) of NUP98-HOXA10hd–transduced and expanded cells, repurified for Lin−Sca-1+c-kit+CD43+Mac1dim phenotype (shown on the left). Error bars indicate the SD of the mean.
In conclusion, isolation of GFP+lin−Sca-1+CD43+(c-kit+)Mac1dim cells from cultures of NUP98-HOXA10hd–transduced cells offers a simple method for obtaining HSCs with increased self-renewal activity in vitro in large numbers and at very high purities. Indeed, we have subsequently established the feasibility of scaling up this expansion and purification strategy to obtain an estimated 1 to 2 × 105 highly purified HSCs at 25% purity from as few as 1000 starting E-SLAM cells.
Factorial analysis of the effect of different conditions on NUP98-HOXA10hd–transduced HSC expansion in vitro
Given evidence that external cues can directly and rapidly modulate HSC self-renewal responses in vitro,34,37 we next asked whether this also would be true for NUP98-HOXA10hd–transduced HSCs. To this end, we performed a 2-level full factorial experimental design to assess the effects of IL-3, IL-6, and SCF and TPO on NUP98-HOXA10hd–transduced HSC expansion in 7-day cultures initiated with lin−Sca-1+CD43+(c-kit+)Mac1dim cells isolated from previous 7-day expansion cultures. Aliquots containing 100 of these cells (estimated to contain 20-25 HSCs; see Figure 4B) were exposed to various combinations of IL-3, IL-6, SCF, and TPO (as shown in supplemental Table 7), and 7 days later the cells were harvested and immunophenotyped. We transplanted 1/10 000 of the cells into 2 irradiated recipients, for each condition tested. The total number of viable lin−Sca-1+CD43+(c-kit+)Mac1dim cells recovered from the cultures and the level of chimerism obtained in the recipients 6 weeks and 4 months after transplantation were analyzed. The results for which at least 1 of these readouts was significant (Figure 5; supplemental Figure 1) revealed remarkably consistent positive and negative trends. Both endpoints indicated positive effects of IL-6 and SCF (P < .01) on the expansion process. In addition, these experiments revealed a positive interaction between IL-6 and SCF (P = .0001). The ability of the lin−Sca-1+CD43+(c-kit+)Mac1dim phenotype to predict the results of functional assays of HSC responses (Figure 5) was further supported by the correlation analysis of the phenotypic and 4-month endpoints (R2 = 0.80, P < .0001).

Growth factor effects on NUP98-HOXA10hd–induced HSC expansion in vitro. Graphic representation of the value of factor effects (βi) divided by the model intercept (β0) based on the prediction equation under “Factorial design experiments.” Three responses (Y) were modeled: the number of transduced cells of Lin−Sca-1+c-kit+CD43+Mac1dim phenotype obtained in 7-day multifactorial expansion cultures (1) and the percentage of transduced cells detected 6 weeks (2) and 4 months (3) after transplantation in peripheral blood of recipients of 1/10 000 of each 7-day multifactorial expansion culture. Interaction effects are identified by the multiplication sign, ×. The factor effects are plotted with respect to growth factor concentrations scaled between −1 (low values) and 1 (high values). The values for each of the 4 factors per condition are presented in supplemental Table 7.
Growth factor effects on NUP98-HOXA10hd–induced HSC expansion in vitro. Graphic representation of the value of factor effects (βi) divided by the model intercept (β0) based on the prediction equation under “Factorial design experiments.” Three responses (Y) were modeled: the number of transduced cells of Lin−Sca-1+c-kit+CD43+Mac1dim phenotype obtained in 7-day multifactorial expansion cultures (1) and the percentage of transduced cells detected 6 weeks (2) and 4 months (3) after transplantation in peripheral blood of recipients of 1/10 000 of each 7-day multifactorial expansion culture. Interaction effects are identified by the multiplication sign, ×. The factor effects are plotted with respect to growth factor concentrations scaled between −1 (low values) and 1 (high values). The values for each of the 4 factors per condition are presented in supplemental Table 7.
NUP98-HOXA10hd induces the in vitro expansion of fetal HSCs
To determine whether the self-renewal promoting properties of NUP98-HOXA10hd extended to HSCs from other stages of ontogeny, we transduced aliquots of 50 or single lin−Sca-1+CD43+(c-kit+)Mac1dim E14.5 fetal liver cells (estimated to be ∼10% pure HSCs27 ) and then cultured them for 8 or 12 days, respectively, in the presence of hematopoietic growth factors before performing LDA transplant assays to determine the number of HSCs present. The results from 2 such experiments indicate a net expansion in HSCs numbers of ∼ 900-fold (Figure 6A). Similarly, results from the clonal analyses showed 3 of 5 clones analyzed (60%) produced ∼ 450 to > 1000 daughter HSCs detectable 14 days later (Figure 6A).

NUP98-HOXA10hd induces the in vitro expansion of fetal HSCs. (A) For bulk cultures, hatched bars indicate the number of total nucleated cells (TNCs) at the beginning (day 0) and at the end (day 10) of culture, after NUP98-HOXA10hd infection and in vitro expansion. Value shown for day 10 is the mean ± SD of results from 2 experiments. Filled bars indicate the HSC content in culture on day 0 and day 10, estimated based on HSC frequency calculated by limiting dilution CRU assay. Values shown are the means ± SEM of results from 2 experiments. For clonal cultures, hatched bars indicate the number of TNCs, after NUP98-HOXA10hd infection and in vitro expansion in 14-day culture, for 5 fetal liver (FL) HSC clones tested. Filled bars indicate the number of HSCs generated by day 14 in 5 clonally expanded cultures of NUP98-HOXA10hd–transduced cells, determined by limiting dilution CRU assay. Error bars represent ± 1 SE. The limiting dilution was not reached for clones 1 and 2 and clones 4 and 5 did not contain HSCs. (B) Schematic of the experimental design followed to investigate in vivo competition capacity of fetal liver and adult bm hematopoietic cells. (C) Proportion of peripheral blood leukocyte contribution by fresh adult and fresh fetal (dark and light gray bar on the left) or day 10 NUP98-HOXA10hd–transduced and expanded adult and fresh fetal (dark and light gray bar on the right) HSCs transplanted (16 weeks earlier) to irradiated mice in competition. Values shown are the means ± SD of results from 8 transplanted recipients that received cells from both sources.
NUP98-HOXA10hd induces the in vitro expansion of fetal HSCs. (A) For bulk cultures, hatched bars indicate the number of total nucleated cells (TNCs) at the beginning (day 0) and at the end (day 10) of culture, after NUP98-HOXA10hd infection and in vitro expansion. Value shown for day 10 is the mean ± SD of results from 2 experiments. Filled bars indicate the HSC content in culture on day 0 and day 10, estimated based on HSC frequency calculated by limiting dilution CRU assay. Values shown are the means ± SEM of results from 2 experiments. For clonal cultures, hatched bars indicate the number of TNCs, after NUP98-HOXA10hd infection and in vitro expansion in 14-day culture, for 5 fetal liver (FL) HSC clones tested. Filled bars indicate the number of HSCs generated by day 14 in 5 clonally expanded cultures of NUP98-HOXA10hd–transduced cells, determined by limiting dilution CRU assay. Error bars represent ± 1 SE. The limiting dilution was not reached for clones 1 and 2 and clones 4 and 5 did not contain HSCs. (B) Schematic of the experimental design followed to investigate in vivo competition capacity of fetal liver and adult bm hematopoietic cells. (C) Proportion of peripheral blood leukocyte contribution by fresh adult and fresh fetal (dark and light gray bar on the left) or day 10 NUP98-HOXA10hd–transduced and expanded adult and fresh fetal (dark and light gray bar on the right) HSCs transplanted (16 weeks earlier) to irradiated mice in competition. Values shown are the means ± SD of results from 8 transplanted recipients that received cells from both sources.
Given that fetal liver HSCs are known to be able to outcompete adult bm in cotransplantation repopulation assays,27,38 we next asked whether this property would be retained by their respective NUP98-HOXA10hd–transduced progeny HSCs. To examine this question, we transplanted groups of irradiated mice with equal numbers of genetically distinguishable fresh HSCs from both sources (∼ 30 of each) or equal numbers of fresh fetal and transduced and in vitro–expanded adult HSCs (Figure 6B). As shown in Figure 6C, fetal liver HSCs contributed to long-term reconstitution ∼ 15 times more effectively than fresh or in vitro–expanded adult HSCs. This is consistent with previous observations that the effects of NUP98-HOXA10hd on the self-renewal activity of HSCs are manifest in vitro but are constrained by the regulatory conditions operative in vivo.
NUP98-HOXA10hd–transduced HSCs that undergo high level expansion in vitro continue to expand in vivo
We next sought to investigate the possible consequences of high-level in vitro expansion of HSCs on their subsequent in vivo self-renewal and regenerative activities. To address this question, the HSC content of 2 representative primary recipients was determined by flow cytometry and secondary LDA of the LSK cells produced (Figure 7A Δ2). Each of these mice had been initially transplanted with ∼ 30 HSCs derived from a single HSC, transduced, and expanded in vitro (supplemental Table 1 clones 7 and 8), resulting in high-level (∼ 60% and ∼ 80%, respectively), long-term (6-7-month) lympho-myeloid reconstitution of the recipient (Figure 7C left FACS plots). The proportion of transduced LSK bm cells in the 2 reconstituted mice was comparable with their counterparts in normal, unmanipulated mice (data not shown). Secondary LDA of bm cells obtained from each of these 2 primary recipients show that the ∼ 30 transduced HSCs injected into the primary recipients had, on average, expanded a further ∼ 300-fold (Figure 7B Δ2) that enabled the HSC compartment of each primary recipient to be reconstituted to up to 70% of its expected or normal level. Moreover, these progeny NUP98-HOXA10hd–transduced HSCs continued to provide significant and balanced reconstitution of the myeloid and lymphoid lineages (Figure 7C middle FACS plots).

Progeny of in vitro–expanded NUP98-HOXA10hd–transduced HSC clones continue to expand in vivo and re-expand in vitro. (A) Schematic of the experimental design followed to measure the level of NUP98-HOXA10hd–induced HSCs expansion during 14-day culture in vitro (Δ1), 7-month regeneration in vivo (Δ2), and 6-day re-expansion culture in vitro (Δ3). (B) Bars represent HSC content at the beginning (light gray) and at the end (dark gray) of each expansion period, measured by 10 or 20 LDA CRU assay. Δ1, Δ2, and Δ3 are estimated based on changes in HSC input versus output numbers. (C) Representative peripheral blood FACS plots of primary and secondary recipients transplanted with the progeny of NUP98-HOXA10hd–expanded HSCs, as indicated in panel A.
Progeny of in vitro–expanded NUP98-HOXA10hd–transduced HSC clones continue to expand in vivo and re-expand in vitro. (A) Schematic of the experimental design followed to measure the level of NUP98-HOXA10hd–induced HSCs expansion during 14-day culture in vitro (Δ1), 7-month regeneration in vivo (Δ2), and 6-day re-expansion culture in vitro (Δ3). (B) Bars represent HSC content at the beginning (light gray) and at the end (dark gray) of each expansion period, measured by 10 or 20 LDA CRU assay. Δ1, Δ2, and Δ3 are estimated based on changes in HSC input versus output numbers. (C) Representative peripheral blood FACS plots of primary and secondary recipients transplanted with the progeny of NUP98-HOXA10hd–expanded HSCs, as indicated in panel A.
We next asked whether the ability of the original NUP98-HOXA10hd–transduced HSCs to expand in vitro would be retained by their progeny HSCs regenerated in vivo. Accordingly, LSK bm cells were isolated from the same primary recipients already investigated (Figure 7A Δ3) and were cultured in vitro for 6 days before being assessed by LDA transplants for the number of HSCs present. Significant cell expansion was obtained with an absence of Lin+ cells, and the secondary limiting dilution transplants revealed a further 100-fold expansion of the NUP98-HOXA10hd–transduced HSCs had occurred (Figure 7B Δ3), with retention of a balanced lympho-myeloid differentiation potential (Figure 7C right FACS plots).
Discussion
In this study, we describe the clonal expansion of highly purified preparations of both adult and fetal HSCs after their transduction with a NUP98-HOXA10hd vector and a strategy for repurifying the derived HSCs at purities of at least 25%.28,29,34 These findings demonstrate the consistency of the high (> 1000-fold) HSC expansions achievable from these cells within a 10- to 14-day culture period. The expanded HSCs also showed sustained lympho-myeloid repopulating ability without evidence of any lineage preference, or any deviation in vivo in their regeneration of a normal-sized HSC compartment. Nevertheless, these HSCs retain an ability to amplify their numbers ex vivo under conditions that are inadequate to support normal HSC self-renewal divisions. These properties suggest an ability of NUP98-HOXA10hd to bypass the dependence of normal HSCs on unknown factors required to sustain their ability to self-renew.
This is intriguing given that recent evidence from clonal analysis of adult bm E-SLAM cells has shown that these cells display variable stable differentiation and self-renewal phenotypes. Specifically, these studies have revealed 4 patterns of differentiation behavior of which 2 (those associated with myeloid potential, with or without lymphoid potential) are associated with durable self-renewal activity.28,29 Our data suggest that all of these HSCs can be converted to display a singular differentiation and self-renewal phenotype. Thus, forced overexpression of NUP98-HOXA10hd in HSCs should provide a valuable tool for probing the controls that are responsible for this heterogeneity in the nontransduced starting cells. The finding that 90% of transduced and in vitro–expanded adult bm HSCs are lin−Sca-1+CD43+(c-kit+)Mac1dim cells and that this allows their prospective isolation at purities of at least 25% is also significant. This phenotype is consistent with that expected of a proliferating HSC population in the cultures.
Interestingly, it is known that normal proliferating HSCs cannot be detected at full efficiency because of their failure to engraft when transiting the S/G2/M phases of the cell cycle.39-43 Thus, the purities determined here probably represent ∼ 2-fold underestimates raising the values closer to 50%. Moreover, the demonstrated ability to apply this approach to larger cultures established the feasibility of using these cells for many genetic and chemical screening applications as well as for additional molecular and biologic characterization studies.
Finally, we found that in general a similar pattern of growth factor effects influenced the output of cultured nonmanipulated44 and NUP98-HOXA10hd–transduced transplantable adult HSCs. Factorial analysis has shown that coincident activation of c-kit (via addition of SCF) and gp130 (via addition of IL-6) significantly sustained NUP98-HOXA10hd–induced HSC expansion in vitro. Moreover, a significant correlation was revealed between distinct methods [number of lin−Sca-1+CD43+(c-kit+)Mac1dim–transduced cells obtained in vitro vs. level of chimerism achieved in vivo] used to detect the HSC activity obtained in 7-day culture under such multifactorial conditions. Interestingly, a phenotypic readout suggested that an activation of MPL receptor (via addition of TPO) has an inhibitory effect (P = .02) on NUP98-HOXA10hd–induced HSC expansion in vitro, seemingly to some extent through negative interactions with SCF (P = .02) and IL-6 (P = .03). Such findings illustrate a potential use of phenotype as an initial screening assay that may reduce the experimental burden compared with long-term assay. Such phenotype-based assay would allow higher throughput screening and optimization of in vitro conditions affecting HSC self-renewal and expansion.
This work provides a strategy for the prospective isolation of large numbers of HSCs with durable self-renewal activity and balanced lympho-myeloid differentiation ability. Nevertheless, the enhanced self-renewal ability they can display in vitro relative to their normal counterparts remains subject to the extrinsic factors to which they are exposed both in vitro and in vivo. Thus, this approach should facilitate future elucidation of how intrinsic and extrinsic regulatory elements interact to determine HSC fate decisions by allowing more precise molecular interrogation of their responses to discrete stimuli. Our findings also emphasize the robustness of NUP98-HOXA10hd to enhance the self-renewal program of HSCs from diverse sources (ie, fetal and adult), thereby offering new opportunities to contrast the molecular features that distinguish and unite these HSCs. Further exploitation of these observations should not only provide novel methods for obtaining clinically relevant HSC numbers ex vivo, but probably also help to develop targeted therapies for cancers arising from inappropriately activated self-renewal pathways.
The online version of the article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Anush Zakaryan, Nicole Hofs, Francis Viel, and William Bowden for expert help and technical assistance; and members of the flow core of the Terry Fox Laboratory and staff of the animal facilities of British Columbia Cancer Agency Research Center.
This work was supported by a Terry Fox Foundation Program Project grant, the National Institutes of Health (R01 HL065430-09), the Canadian Institutes of Health Research (CIHR; Team Grant on Stem Cell Expansion), and the Children's Leukemia Research Association and the British Columbia Cancer Foundation. D.G.K. held scholarships from CIHR and the Michael Smith Foundation for Health Research (MSFHR); V.L. held scholarships from the Natural Sciences and Engineering Research Council of Canada and MSFHR; C.A.H. held scholarships from MSFHR and Scott Paper; and C.L.H. held an MSFHR scholarship.
National Institutes of Health
Authorship
Contribution: S.S., M.G., V.L., and C.A.H. designed and performed the research and analyzed the data; D.G.K. provided purified HSCs; P.R. and A.W. performed viral integration analysis; C.B. helped perform mouse experiments; C.L.H., J.M.P., and C.S. designed the research and edited the manuscript; C.J.E. and R.K.H. designed the research; and S.S., C.J.E., and R.K.H. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: R. Keith Humphries, Terry Fox Laboratory, BC Cancer Agency, 675 West 10th Avenue, Vancouver, BC, Canada V5Z 1L3; e-mail: khumphri@bccrc.ca.
