

Abstract
Interferon-α (IFN-α)–based therapy is presently the standard treatment for hepatitis C virus (HCV)–infected patients. Despite good effectiveness, this cytokine is associated with major side effects, including significant lymphopenia, that limits its use for HIV/HCV-coinfected patients. Interleukin-7 (IL-7) has recently shown therapeutic potential and safety in several clinical trials designed to demonstrate T-cell restoration in immunodeficient patients. The purpose of this study was to evaluate, in simian immunodeficiency virus-infected rhesus macaques, the relevance of IL-7 therapy as a means to overcoming IFN-α–induced lymphopenia. We showed that low-dose IFN-α treatment induced strong lymphopenia in chronically infected monkeys. In contrast, high-dose IFN-α treatment stimulated IL-7 production, leading to increased circulating T-cell counts. Moreover, IL-7 therapy more than abrogated the lymphopenic effect of low-dose IFN-α. Indeed, the association of both cytokines resulted in increased circulating T-cell counts, in particular in the naive compartments, as a consequence of central and peripheral homeostatic functions of the IL-7. Finally, reduced PD-1 expression by memory CD8+ T cells and transient T-cell repertoire diversification were observed under IL-7 therapy. Our data strongly suggest that IL-7 immunotherapy will be of substantial benefit in the treatment of HIV/HCV coinfection and should enhance the likelihood of HCV eradication in poorly responding patients.
Introduction
Coinfection with HIV and hepatitis C virus (HCV) has become an increasingly common occurrence, with an estimated 25% to 30% of HIV+ patients also being HCV+.1 As a consequence, since the appearance of highly active antiretroviral therapy (HAART), chronic liver disease has become one of the most common causes of morbidity and mortality in HIV patients.2,3
Although the effectiveness of standard HCV treatment, based on interferon-α (IFN-α) injections, is known to decrease the longer a patient has been infected with the virus, HCV patients are not systematically put under treatment. Rather, initiation of treatment depends on the state of the patient's liver (fibrosis markers, serum aminotransferases, HCV RNA, hepatitis B virus status) and other parameters, which in turn are used to determine whether the potential benefits of therapy outweigh the risks of treatment-related toxicity.4 For HIV-HCV–coinfected patients, some of these parameters are: the assessment of HIV disease status, such as current and past opportunistic infections, HIV-associated malignancies, CD4 count, HIV viral load, and details of HAART therapy, with CD4 count being one of the main criteria. Unfortunately, coinfected patients are poor responders to standard treatment as a sustained viral response occurs in only 27% to 40% of these patients, approximately half the rate seen in HCV-monoinfected patients.5-7 The main reason for this difference can be attributed to exhaustion of the immune system. In addition, IFN-α treatment causes a leukopenia, including a decrease of lymphocyte numbers and thus of CD4 count, often leading to premature treatment interruption.8-12 Therefore, to enhance the efficiency of anti-HVC therapy in coinfected patients, it is critically important to develop an alternative treatment that limits the lymphopenic effect of IFN-α therapy.
Interleukin 7 (IL-7), a cytokine that promotes precursor B- and T-cell maturation13-15 and supports peripheral T-cell homeostasis,16-18 is currently in phase I/II trials in patients with persistent lymphopenia. This therapy has demonstrated its efficacy in restoring circulating CD4+ T-cell counts in HAART-treated HIV-infected patients19,20 as well as in increasing T-cell diversity in patients under chemotherapy for cancer.21 In healthy as well as in simian immunodeficiency virus (SIV)-infected rhesus macaques under antiretroviral therapy, recombinant glycosylated simian IL-7 (R-sIL-7gly) has been shown to increase peripheral proliferation/survival of mature T cells and to promote both central production of new T cells in the thymus.22,23 Moreover, IL-7 has shown an ability to enhance antigen-specific immune responses in vitro.24,25
Considering the various properties of IL-7 and its high degree of tolerance in patients, this study was designed with the idea of adding recombinant IL-7 to IFN-α–based therapy as a way to limit lymphopenia as well as to enhance antiviral immune responses in HIV-HCV–coinfected patients. The aim of the present study was to evaluate, in the SIV-infected rhesus macaque model, the effectiveness of R-sIL-7gly in maintaining circulating T-cell numbers when coadministered with PEG-IFN-α. Moreover, we evaluated the consequences of this combination treatment on both T-cell diversity and anti-SIV specific immune responses.
Methods
Animals and SIV infection
Nine rhesus macaques, housed and cared for in accordance with the Directive of the European Parliament and of the Council on the Protection of Animals Used for Scientific Purposes, were included in this study. The experiments were approved by the ethics committee of the region Ile de France–Paris I, France. The animals were seronegative for simian T-cell leukemia virus type 1, simian retrovirus type 1 (type D retrovirus), and herpesvirus B. All of the animals were inoculated with animal infectious dose 50 of the pathogenic SIVmac251 isolate (provided by A.-M. Aubertin, Institut National de la Santé et de la Recherche Médicale Unité 544, Strasbourg, France). For all injections and blood draws, the animals were anesthetized with Zolétil (tiletamine/zolasepam; 4.6 mg/kg).
IFN-α and R-sIL-7gly treatments
Four animals received an “IFN-α only treatment” consisting of 10 sub-cutaneous injections, once a week, of either 35 μg (nos. 1 and 2) or 135 μg (nos. 6 and 7) of pegylated human IFN-α-2a (Pegasys, Roche Diagnostics).
Five animals received an “IFN-α + IL-7 treatment” consisting of the same IFN-α therapy (35 μg for animals 3-5 or 135 μg for animals 8 and 9) plus 4 subcutaneous injections, once every 3 weeks, of R-sIL-7gly (80 μg/kg body weight), a dose previously shown to stimulate T-cell expansion in both healthy and SIV-infected macaques.23,26 R-sIL-7gly, produced and provided by Cytheris SA, was injected 3 days after the previous IFN-α injection.
The animals were weighed, temperatures were checked, and blood was sampled (5 mL on ethylenediaminetetraacetic acid) once a week for 3 weeks before the first IFN-α injection, during the treatment, and for 1 month after.
Flow cytometry
For fluorescence-activated cell sorter analysis, 500 μL of whole blood of every blood sample was frozen with 500 μL of 20% dimethyl sulfoxide, fetal calf serum, and stored in liquid nitrogen until the end of follow-up.
Once all the samples were collected, a vial from each time point was thawed in 10% fetal calf serum Ca2+Mg2+-free phosphate-buffered saline (PBS), washed twice, and incubated for 20 minutes with conjugated monoclonal antibodies. For intracellular staining, cells were permeabilized and washed with the Cytofix/Cytoperm Kit (BD Biosciences) according to the manufacturer's instructions. The cells were then incubated for 15 minutes with specific monoclonal antibodies. Samples were washed twice with PBS and fixed in 2% paraformaldehyde PBS. Immunostainings were produced using a Cyan cytofluorometer (Dako North America) and analyzed with FlowJo software, Version 8.8.6 (TreeStar).
The different monoclonal antibodies used in this study were: CD3-allophycocyanin or CD3-phycoerythrin (PE)-Cy7 (clone SP34-2), CD4-peridinin chlorophyll protein-Cy5.5 (clone L200), CD8-Pacific Blue (clone RPA-T8), CD28-PE (clone CD28.2), CD95-allophycocyanin (clone DX2), streptavidin-PE–Texas Red and PD-1–PE (clone MIH4) purchased from BD Biosciences; CD31-biotin (clone WM59) from Serotec; and Ki-67–fluorescein isiothiocyanate (clone MIB-1) and Bcl-2-fluorescein isiothiocyanate (clone 124) from Dako North America.
IL-7 plasma quantification
IL-7 was quantified in the plasma using the IL-7 QuantikineR HS kit according to the manufacturer's instructions (R&D Systems Europe).
TREC quantification
Parallel triplicate quantification of the sjTREC and each of the 13 DJβTRECs, together with CD3γ gene (used as a housekeeping gene) was performed for each sample using LightCycler technology (Roche Diagnostics) as previously described.27 Briefly, peripheral blood mononuclear cells (PBMCs) were lysed in Tween-20 (0.05%), NP-40 (0.05%), and proteinase K (100 μ g/mL) for 30 minutes at 56°C and then 15 minutes at 98°C. Multiplex polymerase chain reaction (PCR) amplification was performed for sjTREC together with the CD3γ chain, in a final volume of 100 μL (10 minutes initial denaturation at 95°C, then 22 cycles of 30 seconds at 95°C, 30 seconds at 60°C, 2 minutes at 72°C) using outer 3′/5′ primer pairs.23 PCR conditions in the LightCycler experiments, performed on 1/100th of the initial PCR products, were: 1 minute initial denaturation at 95°C, then 40 cycles of 1 second at 95°C, 10 seconds at 60°C, and 15 seconds at 72°C. Measurements of the fluorescent signals were performed at the end of annealing steps. TREC and CD3γ LightCycler quantifications were performed in independent experiments, using the same first-round serial dilution standard curve. Similarly, DJβ1TRECs (DJβ1.1-1.6) and DJβ2TRECs (DJβ2.1-2.7) were quantified in multiplex quantitative PCR assays. This highly sensitive nested quantitative PCR assay made it possible to detect one TREC molecule in 105 cells for any excision circle. The sj/βTREC ratio was calculated as previously described:
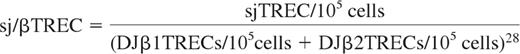
Quantitative immunoscope
Determination of Vβ gene usage and Immunoscope analysis were performed as described by Lim et al.29 In brief, total RNA was extracted from approximately 3 × 106 PBMCs from time points before initiation (day −21), at the end of the treatments (day 63), and from one month after the last injection (day 94), using the GenElute Mammalian Total RNA Miniprep kit (Sigma-Aldrich) according to the manufacturer's specifications. cDNA was then prepared by RNA reverse transcription with 0.5 μg/μL oligo(dT)17 and 200 U of SuperScript II reverse transcriptase (Invitrogen). An aliquot of cDNA synthesis reaction was amplified with each of the 24 T-cell receptor Vβ family–specific primers, together with a T-cell receptor Cβ primer and a TaqMan probe (Applied Biosystems), designed on rhesus macaque germline sequences (GenBank accession number NC 007860; supplemental Table 1, available on the Blood Web site; see the Supplemental Materials link at the top of the online article). Real-time quantitative PCR was conducted in an ABI7300 device (Applied Biosystems). In a second approach, 2 μL of each of these amplification reactions was used as template in run-off reactions using a nested fluorescent primer specific for the Cβ segment. In this reaction, all PCR products were copied into fluorescently labeled single-stranded DNA fragments, irrespective of their T-cell receptor Jβ usage or CDR3 sequence. These fluorescent products were separated on an ABI-PRISM 3730 DNA analyzer (Applied Biosystems). The size and intensity of each band were analyzed with Immunoscope Version 3-6.3 software.30 The diversity of T-cell repertoire in the different samples was analyzed compared with a standard Gaussian distribution obtained from a similar experiment performed on a young healthy rhesus macaque blood sample. T-cell repertoire diversity was calculated as 1/quadratic distance for each sample, calculated as previously described.31
PBMC antigen stimulation and intracellular cytokine staining
PBMCs of 4 of the treated macaques (nos. 6-9) from time points before initiation (day −21), at the end of the treatments (day 56), and from 5 weeks after the last injection (day 102) were thawed and then cultured and stimulated with SIV-peptides. PBMCs were washed once in ambient temperature culture medium (RPMI 10% fetal calf serum) containing 20 μg/mL of DNase I (Sigma-Aldrich) and once in PBS. The cells were then counted and adjusted to 1 × 106 cells/100 μL with DNase I-containing culture medium and distributed through 6 wells of a conic 96-well plate. Then, 100 μL of stimulation media (DNase culture medium; brefeldin A [1/1000 Golgi-plug; BD Biosciences]; anti-CD49a [1 ng/mL; clone SR84, BD Biosciences]; anti-CD28 [1 ng/mL; clone CD28.2, BD Biosciences]; and either 2 μg/mL Staphylococcus aureus enterotoxin B (Sigma-Aldrich) for positive control, dimethyl sulfoxide for negative control, or 5 μg/mL of one of 4 pools of SIV peptides) was added at hour 0. The peptide pools, 3 for Gag protein and 1 for Nef protein, were obtained from the National Institutes of Health. The cells were then incubated for 6 hours at 37°C, 5% CO2. After 6 hours of incubation, the plates were cooled on ice and the cells harvested into ice-cooled tubes and washed with cold PBS.
Cells were then stained as described in “Flow cytometry,” using the following monoclonal antibodies: CD3-PE-Cy7 (clone SP34-2), CD4-peridinin chlorophyll protein-Cy5.5 (clone L200), CD8-Pacific Blue (clone RPA-T8), IFN-γ–allophycocyanin (clone 4S.B3), TNF-α–fluorescein isiothiocyanate (clone Mab11), and IL-2–PE (clone MQ1-17H12), all purchased from BD Biosciences.
Immunostainings were produced using an LSRII (BD Biosciences) and analyzed with FlowJo software, Version 8.8.6 (TreeStar).
Statistical analysis
Statistical analyses (Spearman rank correlations and Wilcoxon matched paired signed-rank tests) were performed using the VassarStats Web site (http://faculty.vassar.edu/lowry/VassarStats.html) and the Stata/IC Version 10.0 for Macintosh statistical software package (Stata).
Results
R-sIL-7gly injection counteracts the lymphopenic effect of IFN-α therapy
To evaluate the relevance of adding R-sIL-7gly treatment to IFN-α–based therapy in HIV-HCV–coinfected patients, a preclinical study in the SIV-infected rhesus macaque model was performed. Five chronically SIV-infected monkeys (1-5) received weekly injections of PEG-IFN-α (35 μg) for a total of 10 weeks. This dose is equal to approximately 3 times the effective dose used to cure HCV infection in humans, adjusted for animal body weight. From day 3 of IFN-α therapy, 3 of these animals (3-5) received R-sIL-7gly, one injection (80 μg/kg) every 3 weeks for a total of 4 injections (days 3, 24, 45, and 66). The follow-up period for these animals lasted for 15 weeks (102 days) after the first IFN-α injection. The animals receiving PEG-IFN-α alone (1 and 2) demonstrated T-cell lymphopenia characterized by a reduction in both CD4+ and CD8+ circulating T-cell counts (631-248 and 106-60 CD4+ T cells/μL between initiation of treatment [day 0] and day 73 in macaques 1 and 2, respectively; 582-290 and 193-69 CD8+ T cells/μL), leading to a 2.2- to 2.1-fold decrease in the number of circulating T cells at day 73 (Figure 1A top panels).

R-sIL-7 therapy eliminates PEG-IFN-α–induced lymphopenia. (A) Evolution of CD3+ (left panels), CD4+ (middle panels), and CD8+ (right panels) T-cell counts in IFN-α and IFN-α plus IL-7–treated macaques. Macaques 1 and 2 (top panels; white and gray bars, respectively) were treated with weekly injections of PEG-IFN-α (35 μg/kg). Macaques 3, 4, and 5 (bottom panels; white, gray, and black bars, respectively) received weekly injections of PEG-IFN-α (35 μg/kg) and 4 injections of R-sIL-7gly (80 μg/kg) once every 3 weeks for a total of 10 weeks. (B) Evolution of CD4+ naive (top panels) and central memory (TCM; bottom panels) T-cell counts in macaques 1 and 2 (left panels; white and gray bars, respectively) as well as in macaques 3, 4, and 5 (right panels; white, gray, and black bars, respectively). (C) Evolution of CD8+ naive (top panels) and central memory (TCM; bottom panels) T-cell counts in macaques 1 and 2 (left panels; white and gray bars, respectively) as well as in macaques 3, 4, and 5 (right panels; white, gray and black bars, respectively).
R-sIL-7 therapy eliminates PEG-IFN-α–induced lymphopenia. (A) Evolution of CD3+ (left panels), CD4+ (middle panels), and CD8+ (right panels) T-cell counts in IFN-α and IFN-α plus IL-7–treated macaques. Macaques 1 and 2 (top panels; white and gray bars, respectively) were treated with weekly injections of PEG-IFN-α (35 μg/kg). Macaques 3, 4, and 5 (bottom panels; white, gray, and black bars, respectively) received weekly injections of PEG-IFN-α (35 μg/kg) and 4 injections of R-sIL-7gly (80 μg/kg) once every 3 weeks for a total of 10 weeks. (B) Evolution of CD4+ naive (top panels) and central memory (TCM; bottom panels) T-cell counts in macaques 1 and 2 (left panels; white and gray bars, respectively) as well as in macaques 3, 4, and 5 (right panels; white, gray, and black bars, respectively). (C) Evolution of CD8+ naive (top panels) and central memory (TCM; bottom panels) T-cell counts in macaques 1 and 2 (left panels; white and gray bars, respectively) as well as in macaques 3, 4, and 5 (right panels; white, gray and black bars, respectively).
In contrast, the animals treated with coinjections of PEG-IFN-α and R-sIL-7gly (3-5) showed a significant increase in circulating CD4+ and CD8+ T-cell counts during therapy, leading to a mean 2.3-fold increase in CD3+ T-cell counts by day 52 (Figure 1A bottom panels), with the maximum circulating T-cell numbers reached after the third R-sIL-7gly injection. After the initial increase in T-cell counts during the period of therapy, circulating CD4+ and CD8+ T-cell counts gradually diminished during the follow-up period but remained at an enhanced level in 2 animals until the end of the study, 5 weeks after the end of therapy.
We then quantified the evolution of naive (CD95− CD28+) and memory (CD95+CD28+) T cells in the PEG-IFN-α alone and PEG-IFN-α plus R-sIL-7gly–treated macaques. Interestingly, whereas IFN-α therapy similarly affected cell counts in all T-cell subsets (1.7- to 2.9-fold decrease in any subset at day 73; Figure 1B-C left panels), the R-sIL-7gly treatment led to preferential enhancement of the naive T-cell populations (3.8- to 6.1-fold increase and 2.0- to 5.1-fold increase for naive CD4+ and CD8+, respectively, at maximum change; Figure 1B-C top right panels). These data demonstrate that R-sIL-7gly treatment eliminates the lymphopenic effect of IFN-α therapy in SIV-infected rhesus macaques.
R-sIL-7gly enhances naive T-cell proliferation and survival in IFN-α–treated macaques
We further investigated the mechanisms by which R-sIL-7gly counteracts IFN-induced lymphopenia and the consequences of R-sIL-7gly therapy on IFN-α–induced modification of circulating T-cell homeostasis (Figure 2). Cell cycling, estimated by the expression of Ki-67, was strongly affected by the IFN-α therapy, leading to a 5.5- to 14.7-fold decrease and to a 3.6- to 4.5-fold decrease by day 102 in the number of Ki-67-expressing CD4+ and CD8+ T cells, respectively (Figure 2A left panels). These changes in cycling cell counts were a consequence of both total circulating cell count decline and a reduction in the percentage of cycling cells (2.87% to 0.38% and 8.3% to 2.5% of the CD4+ T cells and 2.6% to 0.9% and 7.7% to 2.9% of the CD8+ T cells, for macaques 1 and 2, respectively). In contrast, despite IFN-α therapy, the animals that also received R-sIL-7gly injections maintained the frequency of Ki-67-expressing cells, leading, while total T-cell counts increase, to an enhancement of cycling T-cell numbers (Figure 2A right panels). Of note, waves of Ki-67 expression were observed, occurring approximately one week after each R-sIL-7gly injection. Similarly, R-sIL-7gly injections also stimulated waves of overexpression of the antiapoptotic molecule Bcl-2 in both CD4+ and CD8+ T-cell subsets (Figure 2B right panels). Interestingly, the evolution of naive T-cell cycling (Figure 2C) and survival (Figure 2D) paralleled that of the CD4+ and CD8+ subsets (Figure 2A-B), suggesting that the increase in circulating T-cell numbers in monkeys treated with combination therapy was, at least in part, the result of recurrent enhancement of T-cell survival and cycling.

R-sIL-7gly induces cell cycling and cell survival in IFN-α–treated macaques. (A) Cell cycling estimated through Ki-67 expression in CD4+ (top panels) and CD8+ (bottom panels) in macaques 1 and 2 treated with weekly injections of PEG-IFN-α (35 μg/kg) for 10 weeks (left panels; white and gray bars, respectively) as well as in macaques 3, 4, and 5 (right panels; white, gray, and black bars, respectively) that received weekly injections of PEG-IFN-α (35 μg/kg) and 4 injections of R-sIL-7gly (80 μg/kg) once every 3 weeks for a total of 10 weeks. (B) Bcl-2 expression in CD4+ (top panels) and CD8+ (bottom panels) for macaques 1 and 2 (left panels; white circles and squares, respectively) and macaques 3, 4, and 5 (right panels; gray diamonds, triangles, and squares, respectively). (C) Ki-67 expression in naive CD4+ (top panels) and CD8+ (bottom panels) T cells for macaques 1 and 2 (left panels; white and gray bars, respectively) and macaques 3, 4, and 5 (right panels; white, gray, and black bars, respectively). (D) Bcl-2 expression in naive CD4+ (top panels) and CD8+ (bottom panels) T cells for macaques 1 and 2 (left panels; white circles and squares, respectively) and macaques 3, 4, and 5 (right panels; gray diamonds, triangles, and squares, respectively). (E) Plasma viral load measured throughout the follow-up period for macaques 1 and 2 (left panels; white circles and squares, respectively) and macaques 3, 4, and 5 (right panels; gray diamonds, triangles, and squares, respectively).
R-sIL-7gly induces cell cycling and cell survival in IFN-α–treated macaques. (A) Cell cycling estimated through Ki-67 expression in CD4+ (top panels) and CD8+ (bottom panels) in macaques 1 and 2 treated with weekly injections of PEG-IFN-α (35 μg/kg) for 10 weeks (left panels; white and gray bars, respectively) as well as in macaques 3, 4, and 5 (right panels; white, gray, and black bars, respectively) that received weekly injections of PEG-IFN-α (35 μg/kg) and 4 injections of R-sIL-7gly (80 μg/kg) once every 3 weeks for a total of 10 weeks. (B) Bcl-2 expression in CD4+ (top panels) and CD8+ (bottom panels) for macaques 1 and 2 (left panels; white circles and squares, respectively) and macaques 3, 4, and 5 (right panels; gray diamonds, triangles, and squares, respectively). (C) Ki-67 expression in naive CD4+ (top panels) and CD8+ (bottom panels) T cells for macaques 1 and 2 (left panels; white and gray bars, respectively) and macaques 3, 4, and 5 (right panels; white, gray, and black bars, respectively). (D) Bcl-2 expression in naive CD4+ (top panels) and CD8+ (bottom panels) T cells for macaques 1 and 2 (left panels; white circles and squares, respectively) and macaques 3, 4, and 5 (right panels; gray diamonds, triangles, and squares, respectively). (E) Plasma viral load measured throughout the follow-up period for macaques 1 and 2 (left panels; white circles and squares, respectively) and macaques 3, 4, and 5 (right panels; gray diamonds, triangles, and squares, respectively).
As expected, IFN-α therapy and R-sIL-7gly plus IFN-α coadministration did not significantly affect plasma viral loads (Figure 2E). Indeed, viral loads remained stable in both IFN-α–treated monkeys as well as in macaque 5. In contrast, macaques 3 and 4 showed fluctuations in their viral load, with an overall 2- to 10-fold increase over the follow-up period.
R-sIL-7gly therapy counteracts the negative effect of IFN-α therapy on thymic function
We further analyzed the evolution of the naive T-cell compartment by quantifying recent thymic emigrants (RTEs), as defined by high expression of CD31 (CD31Hi) on naive T cells, and through quantification of the sjTREC molecule in peripheral blood cells. In both IFN-α–treated monkeys, the sjTREC concentration (sjTREC/μL; Figure 3A left panel), the absolute number of RTE CD4+ T cells (Figure 3B top left panel), and, to a lesser extent, CD31low naive T-cell counts (Figure 3B bottom left panel) decreased over time. Of note, CD31− naive T-cell counts were barely affected by PEG-IFN-α therapy in these animals (data not shown). In contrast, the addition of R-sIL-7gly to PEG-IFN-α therapy led to an increase in sjTREC concentrations during the period of therapy in all 3 treated macaques (Figure 3A right panel). Similarly, in one of the PEG-IFN-α plus R-sIL-7gly-treated animals, RTE CD4 T-cell counts increased (Figure 3B top right panel). In the other 2 monkeys, the number of CD4+ RTEs remained stable throughout the follow-up period (Figure 3B top right panel). The stability/increase of sjTREC levels and of RTE CD4+ T-cell counts may suggest that an enhancement of thymic output contributed to the observed increase in naive T-cell counts in these animals. However, whereas IFN-α therapy principally affected the RTE subset, with relative stability observed in the other naive T-cell subsets, RTEs produced under R-sIL-7gly treatment seemed to rapidly mature into naive T cells characterized by a lower CD31 expression (Figure 3B bottom right panel).

R-sIL-7gly stimulates thymic function in PEG-IFN-α–treated macaques. (A) sjTREC blood concentration measured in macaques 1 and 2 treated with weekly injections of PEG-IFN-α (35 μg/kg) for 10 weeks (left panels; white circles and squares, respectively) as well as in macaques 3, 4, and 5 (right panels; gray diamonds, triangles, and squares, respectively) that received weekly injections of PEG-IFN-α (35 μg/kg) and 4 injections of R-sIL-7gly (80 μg/kg) once every 3 weeks for a total of 10 weeks. (B) CD31Hi CD4+ (RTE CD4+; top panels) and CD31Low (bottom panels) T-cell counts measured in macaques 1 and 2 (left panels; white and gray bars, respectively) and in macaques 3, 4, and 5 (right panels; white, gray, and black bars, respectively). Right y-axes correspond to macaques 2 (top and bottom left panels) and 5 (bottom right panel).
R-sIL-7gly stimulates thymic function in PEG-IFN-α–treated macaques. (A) sjTREC blood concentration measured in macaques 1 and 2 treated with weekly injections of PEG-IFN-α (35 μg/kg) for 10 weeks (left panels; white circles and squares, respectively) as well as in macaques 3, 4, and 5 (right panels; gray diamonds, triangles, and squares, respectively) that received weekly injections of PEG-IFN-α (35 μg/kg) and 4 injections of R-sIL-7gly (80 μg/kg) once every 3 weeks for a total of 10 weeks. (B) CD31Hi CD4+ (RTE CD4+; top panels) and CD31Low (bottom panels) T-cell counts measured in macaques 1 and 2 (left panels; white and gray bars, respectively) and in macaques 3, 4, and 5 (right panels; white, gray, and black bars, respectively). Right y-axes correspond to macaques 2 (top and bottom left panels) and 5 (bottom right panel).
High-dose IFN-α treatment induces IL-7 production in SIV-infected rhesus macaques
In a second group of macaques, we modified the dosing of PEG-IFN-α therapy to adjust IFN-α-injected doses to the skin surface of the animals. Accordingly, 4 SIV-infected rhesus macaques were injected weekly with 135 μg of PEG-IFN-α (6-9) for a 10-week period. As in the first experiment, 2 of these animals (8 and 9) also received R-sIL-7gly injections (80 μg/kg of body weight) once every 3 weeks for a total of 4 injections (days 3, 24, 45, and 66). Injections of R-sIL-7gly led to increased circulating T-cell counts over the period of therapy despite injections of high doses of PEG-IFN-α (Figure 4A). Of note, whereas macaque 9 demonstrated sustained T-cell increase for at least a month after the end of the therapy period, the response of macaque 8 was less sustained, with its T-cell counts rapidly returning to baseline levels at the end of the R-sIL-7gly therapy. As it turned out, this animal was ultimately categorized as a rapid progressor.

High-dose IFN-α therapy induces increased T-cell counts. Circulating blood CD3+ (left panels), CD4+ (middle panels), and CD8+ (right panels) T-cell counts measured (A) in macaques 8 and 9 (top panels; black diamonds and triangles, respectively), treated with weekly injections of PEG-IFN-α alone (135 μg/kg) and 4 injections of R-sIL-7gly (80 μg/kg) once every 3 weeks, for a total of 10 weeks and (B) in macaques 6 and 7 (bottom panels; white diamonds and triangles, respectively) treated with weekly injections of PEG-IFN-α (135 μg/kg) for a total of 10 weeks.
High-dose IFN-α therapy induces increased T-cell counts. Circulating blood CD3+ (left panels), CD4+ (middle panels), and CD8+ (right panels) T-cell counts measured (A) in macaques 8 and 9 (top panels; black diamonds and triangles, respectively), treated with weekly injections of PEG-IFN-α alone (135 μg/kg) and 4 injections of R-sIL-7gly (80 μg/kg) once every 3 weeks, for a total of 10 weeks and (B) in macaques 6 and 7 (bottom panels; white diamonds and triangles, respectively) treated with weekly injections of PEG-IFN-α (135 μg/kg) for a total of 10 weeks.
The animals that received the higher doses of PEG-IFN-α alone showed waves of increases of circulating T-cell numbers over the treatment period (1.5- and 2.6-fold increase in CD3+ T-cell number, 2.3- and 3.3-fold increase in CD4+ T-cell number and 1.2- and 2.5-fold increase in CD8+ T-cell number, respectively, in macaques 6 and 7, at maximum increase; Figure 4B). However, this effect was short-lived as T-cell counts returned to baseline levels by day 70 in both animals.
We then further analyzed the regulation of T-cell homeostasis in the animals that received the higher dose of PEG-IFN-α alone. Along with the general decrease of Ki-67 expression observed throughout the period of treatment, waves of cell cycling were observed in the CD4+ T-cell compartment at days 14 and 42 (Figure 5A). Moreover, unlike in the animals receiving the low dose of PEG-IFN-α, Bcl-2 expression was also enhanced during the therapy period (Figure 5B). Surprisingly, both animals that received the higher dose of PEG-IFN-α alone reacted in the first weeks of therapy with increased IL-7 production (Figure 5C). Indeed, IL-7 plasma levels reached a first maximum during days 14 to 21 (initial plasma level: ∼ 14 pg/mL [8.6-15.2 pg/mL], maximal plasma concentration: ∼ 61 pg/mL [27-71pg/mL]; Figure 5C). Of note, macaques 1 and 2 did not react to the low dose of IFN-α by producing IL-7 (Figure 5C). Thereafter, IL-7 plasma levels oscillated with peaks of production every 2 to 3 weeks. In the 2 macaques treated with high doses of PEG-IFN-α, waves of IL-7 production coincided with variations in both naive CD4+ and CD8+ T-cell counts (Figure 5D) as well as with variations in sjTREC concentrations (Figure 5E), suggesting that IL-7 produced in response to high-dose PEG-IFN-α therapy also positively impacted thymopoiesis. Indeed, in these 2 animals, sjTREC blood concentrations were directly correlated with total (CD4+ + CD8+) naive T-cell counts (Figure 5F).

Consequences of high-dose IFN-α therapy. Cell cycling estimated through Ki-67 expression (A); cell survival estimated through Bcl-2 expression (B) and naive T-cell counts (D) in CD4+ (top panels) and CD8+ (bottom panels) from macaques 6 and 7 (diamonds and triangles, respectively) treated with weekly injections of PEG-IFN-α (135 μg/kg) for a total of 10 weeks. (C) Plasma IL-7 levels quantified in macaques 1, 2, 6, and 7 (circles, squares, diamonds, and triangles, respectively). (E) sjTREC concentration quantified in macaques 6 and 7 (diamonds and triangles, respectively). (F) Relationships between naive T-cell counts and sjTREC concentrations in macaques 6 and 7 (diamonds and triangles, respectively).
Consequences of high-dose IFN-α therapy. Cell cycling estimated through Ki-67 expression (A); cell survival estimated through Bcl-2 expression (B) and naive T-cell counts (D) in CD4+ (top panels) and CD8+ (bottom panels) from macaques 6 and 7 (diamonds and triangles, respectively) treated with weekly injections of PEG-IFN-α (135 μg/kg) for a total of 10 weeks. (C) Plasma IL-7 levels quantified in macaques 1, 2, 6, and 7 (circles, squares, diamonds, and triangles, respectively). (E) sjTREC concentration quantified in macaques 6 and 7 (diamonds and triangles, respectively). (F) Relationships between naive T-cell counts and sjTREC concentrations in macaques 6 and 7 (diamonds and triangles, respectively).
These data show that treatment with the higher dose of PEG-IFN-α stimulated endogenous IL-7 production at a level sufficient to counteract the lymphopenic effect of IFN-α through increased peripheral T-cell proliferation and survival and probably through enhanced thymic function, although this naturally occurring protection only lasted approximately a month.
R-sIL-7gly therapy leads to transient diversification of T-cell repertoire
Taking into consideration that R-sIl-7gly therapy potentially enhanced thymic function, we next analyzed T-cell repertoire diversity in these animals, hypothesizing that the newly produced T cells and the proliferation of naive T cells would increase T-cell polyclonality in the chronically SIV-infected, non–HAART-treated macaques. We first quantified the extent of intrathymic precursor T-cell proliferation through calculation of the sj/βTREC ratio in 3 animals injected with either low or high doses of PEG-IFN-α as well as in the macaques treated with high doses of PEG-IFN-α plus R-sIL-7gly. This parameter, which is strongly affected in HIV- and SIV-infected persons, significantly contributes to the establishment of T-cell repertoire diversity.32 As expected for SIV-infected macaques, baseline sj/βTREC ratios were low (median sj/βTREC ratio for the 5 animals at baseline = 19; range, 4-109). Moreover, monkeys treated with PEG-IFN-α alone (low and high dose) did not show any significant changes over the follow-up period (Figure 6A left and middle panels). Of note, the sj/βTREC ratio could not be calculated for macaque 1 because of very low sjTREC levels and undetectable DJβTRECs. In contrast, both PEG-IFN-α plus R-sIL-7gly–cotreated monkeys demonstrated a strong increase in the sj/βTREC ratio with a maximum reached by days 14 to 21 (Figure 6A right panel), confirming the impact of R-sIL-7gly on thymic function. In both macaques, this effect was only transient, with the sj/βTREC ratio returning to baseline levels by the end of the treatment period (Figure 6A right panel).

Evolution of T-cell diversity in PEG-IFN-α– and R-sIL-7gly–treated macaques. (A) Evolution of the sj/βTREC ratio measured in macaques 1 and 2 (left panel; white circles and squares, respectively) treated with weekly injections of PEG-IFN-α (35 μg/kg) for a total of 10 weeks, macaques 6 and 7 (central panel; white diamonds and triangles, respectively) treated with weekly injections of PEG-IFN-α (135 μg/kg) for a total of 10 weeks, and macaques 8 and 9 (right panel; black diamonds and triangles, respectively) treated with weekly injections of PEG-IFN-α (135 μg/kg) and R-sIL-7gly (80 μg/kg) every 3 weeks for a total of 10 weeks. (B) T-cell repertoire diversity estimated as 1/quadratic distances to a healthy control monkey in macaques 1 and 2 (left panel; white circles and squares, respectively), macaques 6 and 7 (middle panel; white diamonds and triangles, respectively), and macaques 8 and 9 (right panel; black diamonds and triangles, respectively).
Evolution of T-cell diversity in PEG-IFN-α– and R-sIL-7gly–treated macaques. (A) Evolution of the sj/βTREC ratio measured in macaques 1 and 2 (left panel; white circles and squares, respectively) treated with weekly injections of PEG-IFN-α (35 μg/kg) for a total of 10 weeks, macaques 6 and 7 (central panel; white diamonds and triangles, respectively) treated with weekly injections of PEG-IFN-α (135 μg/kg) for a total of 10 weeks, and macaques 8 and 9 (right panel; black diamonds and triangles, respectively) treated with weekly injections of PEG-IFN-α (135 μg/kg) and R-sIL-7gly (80 μg/kg) every 3 weeks for a total of 10 weeks. (B) T-cell repertoire diversity estimated as 1/quadratic distances to a healthy control monkey in macaques 1 and 2 (left panel; white circles and squares, respectively), macaques 6 and 7 (middle panel; white diamonds and triangles, respectively), and macaques 8 and 9 (right panel; black diamonds and triangles, respectively).
We further analyzed T-cell repertoire diversity using Immunoscope technology performed on peripheral blood cells sampled at baseline, at the end of the treatment period (day 63), and a month later (day 94). Looking at the Immunoscope profiles, a diversification of T-cell repertoire appeared to characterize the PEG-IFN-α plus R-sIL-7gly–treated macaques at day 63 (supplemental Figure 1). We quantified this diversification by calculating the quadratic distances of the T-cell repertoire of each sample against a normal repertoire derived from a healthy macaque. T-cell repertoire diversity was calculated as 1/quadratic distance for each sample (Figure 6B). At baseline, T-cell repertoire diversity was variable in the SIV-infected macaques. However, although it remained quite stable in those animals treated with IFN-α alone (Figure 6B left and middle panels), a transient increase in T-cell diversity was observed at day 63 in both monkeys that received PEG-IFN-α plus R-sIL-7gly (Figure 6B right panel). Subsequent to the treatment period, T-cell repertoire diversity dropped in one of these 2 animals (animal 8) but remained over baseline in the other (animal 9). Of note, animal 8 was the poor responder macaque as defined by CD3+, CD4+, and CD8+ T-cell changes under therapy (Figure 4A).
These analyses demonstrated that, despite PEG-IFN-α treatment, R-sIL-7gly therapy-induced increase in thymic function, evidenced through quantification of the sj/βTREC ratio, translated into diversification of the T-cell repertoire.
R-sIl-7gly therapy allows qualitative improvement of SIV-specific immune responses
The increase of repertoire diversity led us to analyze specific immune responses. In doing so, we analyzed the evolution of SIV-specific immune responses in the macaques that received the higher dose of PEG-IFN-α alone or in combination with R-sIL-7gly. The evolution of cell numbers and PD-1 expression in the CD8+ memory compartment were measured by fluorescence-activated cell sorter. The animals that received the higher dose of PEG-IFN-α alone demonstrated either a transient increase or a regular decrease in total CD8+ central memory T cells (TCM; Figure 7A top panel). In contrast, the addition of R-sIL-7gly to the high-dose IFN-α therapy led to an increase of CD8+ TCM counts in both macaques during the treatment period, an increase that persisted during the after month in the “good responder” macaque (Figure 7A bot-tom panel). Similar to the low-dose IFN-α experiments, injections of a high dose of IFN-α, alone or in combination with R-sIL-7gly, did not significantly affect plasma viral loads (Figure 7B).

Evolution of anti-SIV specific immune responses in PEG-IFN-α– and R-sIL-7gly–treated macaques. (A) CD8+ TCM cell counts measured in macaques 6 and 7 (top panel; gray and black bars, respectively) treated with weekly injections of PEG-IFN-α (135 μg/kg) for a total of 10 weeks and macaques 8 and 9 (bottom panel; gray and black bars, respectively) treated with weekly injections of PEG-IFN-α (135 μg/kg) and 4 injections of R-sIL-7gly (80 μg/kg) once every 3 weeks for a total of 10 weeks. (B) Plasma viral load measured throughout the follow-up period in macaques 6 and 7 (top panel; white diamonds and triangles, respectively) and macaques 8 and 9 (bottom panels; black diamonds and triangles, respectively). (C) Percentage of PD-1–expressing CD8+ TCM cells in macaques 6 and 7 (top panel; white diamonds and triangles, respectively) and macaques 8 and 9 (bottom panels; black diamonds and triangles, respectively).
Evolution of anti-SIV specific immune responses in PEG-IFN-α– and R-sIL-7gly–treated macaques. (A) CD8+ TCM cell counts measured in macaques 6 and 7 (top panel; gray and black bars, respectively) treated with weekly injections of PEG-IFN-α (135 μg/kg) for a total of 10 weeks and macaques 8 and 9 (bottom panel; gray and black bars, respectively) treated with weekly injections of PEG-IFN-α (135 μg/kg) and 4 injections of R-sIL-7gly (80 μg/kg) once every 3 weeks for a total of 10 weeks. (B) Plasma viral load measured throughout the follow-up period in macaques 6 and 7 (top panel; white diamonds and triangles, respectively) and macaques 8 and 9 (bottom panels; black diamonds and triangles, respectively). (C) Percentage of PD-1–expressing CD8+ TCM cells in macaques 6 and 7 (top panel; white diamonds and triangles, respectively) and macaques 8 and 9 (bottom panels; black diamonds and triangles, respectively).
The frequency of PD-1-expressing cells in the CD8+ TCM subset also evolved during IFN-α therapy (Figure 7C top panel). In all 4 macaques, we observed an overall decline in the frequency of PD-1+ CD8+ TCM during and after the period of treatment. However, in the R-sIL-7gly–treated monkeys, each injection of IL-7 was immediately followed by a transient drop in the percentage of PD-1–expressing cells, suggesting an improved antiviral activity (Figure 7C bottom panel). Interestingly, contrary to IFN-α therapy that did not qualitatively impact on the SIV-specific immune response, IL-7–based therapy led to increased frequency and numbers of polyfunctional anti-SIV CD8+ T cells (supple-mental data).
Discussion
Over the last decade, immunotherapy has become increasingly important in the treatment of human pathologies. IFN-α–based therapy was the first and remains one of the most used treatments for viral infections, such as hepatitis C33 as well as an immunomodulating compound for stimulation of specific immunity in advanced cancers.34-37 Despite its efficacy, IFN-α treatment is always associated with significant side effects. Among these, the development of lymphopenia often limits its use, particularly in those patients who have received aggressive chemotherapy or who have T-cell depletion, such as that associated with chronic HIV infection.9,11,12,38 On the other hand, the administration of recombinant IL-7 in several recent phase I/II trials in either cancer patients after aggressive chemotherapy or in HIV-infected patients under HAART19-21 demonstrated good tolerance and high efficiency. Recombinant glycosylated simian IL-7 therapy elicited a significant and prolonged increase in circulating T-cell numbers, increased thymic output, and diversification of T-cell repertoire.19-21 In the study discussed here, we have shown that, in SIV-infected rhesus macaques, the administration of R-sIL-7gly during IFN-α therapy eliminates the lymphopenic effect of IFN-α, allowing the monkeys to maintain their circulating CD4+ and CD8+ T-cell counts during the period of therapy. Of note is the fact that maintenance of high CD4+ T-cell counts, an important parameter for ensuring the efficacy of IFN-α therapy, is absolutely critical in HIV/HCV–coinfected patients.39,40
Increased T-cell diversity and enhanced functionality of SIV-specific CD8+ T cells were observed in HCV-infected patients successfully treated with PEG-IFN-α during early primary infection.41 In these patients, viral clearance was associated with the rescue of polyfunctional T-cell responses. In contrast, nonvirologic responder patients did not exhibit improved anti-HCV immune responses, showing that the development of HCV-specific immune responses is key to develop and maintain full efficacy of IFN-α therapy.41 Such an effect may be of particular importance in coinfected patients who, because of HIV-infection, have a reduced capacity to develop such immune responses. Of particular note is the fact that high-dose IFN-α therapy allowed significant and durable reduction in the frequency of relapse as well as increased survival in patients treated for high-risk melanoma, as a consequence, at least in part, of increased dendritic and T-cell functions.42-44
In our study, treatment with IFN-α alone did not affect the efficacy of SIV-specific CD8+ T cells. However, the animals were in chronic infection and were not under antiretroviral therapy. In contrast, despite high viral replication, the combination of R-sIL-7gly treatment with IFN-α therapy resulted in decreased PD-1 expression by memory CD8+ T cells and enhanced proportion of polyfunctional CD8+ T cells. Nevertheless, the effect of IL-7 therapy was transient, a probable consequence of the absence of antiretroviral therapy in our study. Considering the very good tolerance associated with IL-7 therapy, repeated cycles of IL-7 treatment might well be beneficial to patients. Moreover, in HIV-HCV–coinfected patients under efficient HAART, it seems reasonable to expect that the addition of recombinant IL-7 to IFN-α therapy will facilitate long-term recovery of HCV-specific cytotoxic T lymphocyte responses. The fact that recombinant IL-7 therapy is associated with long-term CD4 T-cell increase in 2 published clinical trials performed on HIV-infected patients tends to support this conclusion.19,20
One of the most surprising observations in our study was the induction of IL-7 production in the macaques that received the higher doses of PEG-IFN-α. In these nonlymphopenic animals, synchronized waves of IL-7 production were observed during the period of therapy and continued thereafter for at least one month. These waves induced overexpression of Bcl-2 by circulating T cells but were insufficient to induce recurrent cell proliferation. Such a dichotomized effect resembles that observed in vitro where low doses of R-sIL-7gly only enhanced T-cell survival, whereas high doses also led to proliferation.45 The factors triggering IL-7 production in the high-dose IFN-α–treated monkeys remain unclear. Whereas the IL-7 receptor has been described as an IFN-α responding factor, it is not the case for IL-7.46 However, it is possible that transcription of the IL-7 gene is indirectly stimulated by IFN-α. Indeed, type 1 IFN production induced by TLR engagement stimulates IL-7 production by hepatocytes in a model of humanized mice, suggesting that IL-7 production might be an indirect consequence of viral replication.47 Such data can also be put together with IL-7 production in SIV- and HIV-infected persons, who, at the peak of viral production and before the establishment of the initial lymphopenia, usually demonstrate increased IL-7 plasma levels.48 Exploring the feedback mechanisms involved in these cascades might help in better understanding this observation.
In monkeys treated with the low dose of PEG-IFN-α, we observed a rapid decline in the frequency of circulating RTEs, accompanied by a reduction in the concentration of sjTREC molecules in the blood. These data strongly suggest that, during the first month of therapy, IFN-α directly affects thymopoiesis. Such a decrease in thymic production is reminiscent of the thymic defect observed during the first months of HIV infection.28 In that case, we attributed the impairment of thymic function to an inhibition of double-positive thymocyte proliferation to the cytokine storm characterizing acute infection.28 Here, in SIV-infected monkeys, the sj/βTREC ratio was already strongly reduced at the beginning of the IFN-α therapy, precluding further decline (Figure 6). However, it is possible that injection of IFN-α not only impacts the proliferation of double-positive thymocytes but also of other thymocyte subsets that cannot be estimated through measurement of the sj/βTREC ratio, leading to the reduction in the number of cells reaching positive and negative selections, and thus leaving the thymus.
In conclusion, using the SIV-infected rhesus macaque model, we have demonstrated that the addition of R-sIL-7gly therapy to IFN-α–based treatment eliminates IFN-α-induced lymphopenia. Moreover, through both central and peripheral effects, this cytokine allows a significant and sustained increase in circulating T-cell counts despite IFN-α treatment. Finally, IL-7 therapy was shown to enhance T-cell diversity and anti-SIV polyfunctional T-cell responses. Altogether, these data suggest that the addition of recombinant human glycosylated IL-7 to the standard IFN-α/ribavirin therapy in HIV/HCV–coinfected patients may be the key to an effective combination therapy, resulting in viral eradication through enhancement of circulating T-cell counts and stimulation of anti-HCV-specific cytotoxic T lymphocyte responses.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Richard Keatinge for his valuable help in preparing the manuscript and Anne-Sophie Beignon for providing SEB and SIV peptides.
This work was supported by Institut Pasteur, Cytheris S.A., and the Agence Nationale de Recherches sur le SIDA et les hepatites virales. S.B. and V.F.-M. were the recipients of Agence Nationale de Recherches sur le SIDA et les hepatites virales postdoctoral grants.
This work was carried out in partial fulfillment of the doctoral thesis of R.P. at the Université Paris 6.
Authorship
Contribution: R.P., S.B., S.R., J.D., V.F.-M., M.R., B.L., and A.L. performed research; B.A., I.R., and M.M. contributed vital new reagents; C.G. participated in animal care; R.C. and M.M. designed research; and R.C. and R.P. analyzed data and wrote the manuscript.
Conflict-of-interest disclosure: M.M. is the CEO and Founder of Cytheris. B.A., S.B., I.R., and S.R. are employees of Cytheris. Cytheris, which is developing recombinant IL-7 as an immune enhancer, partly supported this work and provided the R-sIL-7gly. The remaining authors declare no competing financial interests.
Correspondence: Rémi Cheynier, Département de Virologie, Institut Pasteur, 25-28, Rue du Docteur Roux 75724, Paris, Cedex 15, France; e-mail: remi.cheynier@inserm.fr.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal