

Abstract
T-cell expression of programmed death receptor-1 (PD-1) down-regulates the immune response against malignancy by interacting with cognate ligands (eg, PD-L1) on tumor cells; however, little is known regarding PD-1 and natural killer (NK) cells. NK cells exert cytotoxicity against multiple myeloma (MM), an effect enhanced through novel therapies. We show that NK cells from MM patients express PD-1 whereas normal NK cells do not and confirm PD-L1 on primary MM cells. Engagement of PD-1 with PD-L1 should down-modulate the NK-cell versus MM effect. We demonstrate that CT-011, a novel anti–PD-1 antibody, enhances human NK-cell function against autologous, primary MM cells, seemingly through effects on NK-cell trafficking, immune complex formation with MM cells, and cytotoxicity specifically toward PD-L1+ MM tumor cells but not normal cells. We show that lenalidomide down-regulates PD-L1 on primary MM cells and may augment CT-011's enhancement of NK-cell function against MM. We demonstrate a role for the PD-1/PD-L1 signaling axis in the NK-cell immune response against MM and a role for CT-011 in enhancing the NK-cell versus MM effect. A phase 2 clinical trial of CT-011 in combination with lenalidomide for patients with MM should be considered.
Introduction
Natural killer (NK) cells are CD56+CD3− large granular lymphocytes that comprise a key cellular compartment of the innate immune system. NK cells have been shown to exert antitumor activity against the malignant plasma cell clone in multiple myeloma (MM).1-4 However, through several established mechanisms, the NK-cell versus MM effect is attenuated as the disease inexorably progresses.5-9 MM is increasing in incidence and remains incurable despite the advent of potent novel therapies such as lenalidomide and bortezomib.10 In fact, both lenalidomide and bortezomib have been shown to confer anti-MM activity, in part, through recovery or enhancement of the NK-cell versus MM effect.11,12
The NK-cell versus MM effect is subject to modulation through intracellular signal transduction cascades initiated by activating and inhibitory receptors at the NK-cell surface interacting with ligands expressed on MM tumor cells. Programmed death 1 (PD-1), a member of the B7 family of cosignaling molecules, and its associated ligands PD-L1 and PD-L2 have been shown to play a key role in down-regulating the T-cell immune response.13 The constitutive or inducible expression of PD-1 has been characterized on several immune cell subsets, including T, B, and dendritic cells; however, to date, comparatively little is known regarding PD-1 expression on NK cells and whether or not the PD-1/PD-L1 axis is involved in the NK-cell versus MM effect.14
CT-011 (CureTech, LTD; previously CT-AcTibody or BAT) is a novel immunoglobulin G1 (IgG1) humanized monoclonal antibody (mAb) that modulates the immune response through interaction with PD-1, with previously demonstrated antitumor efficacy in experimental models of both solid and liquid tumors.15-17 Several human malignancies, including MM, express cognate ligands for PD-1 (eg, PD-L1) and play a key role in tumor immunoevasion.18,19 In a phase 1 clinical trial of patients with advanced hematologic malignancies including MM, CT-011 was demonstrated to be safe and well tolerated with evidence of single-agent clinical beneficial responses in 33% of the patients.20 Given the results of this phase 1 study and the potential complementary mechanisms of action between CT-011 and lenalidomide, we hypothesized these agents in combination may represent a promising novel therapy for MM.
Lenalidomide (Revlimid; Celgene) exerts efficacy in part through enhancement of the NK-cell versus MM effect,11 an effect likely mediated through T-cell production of interleukin-2 (IL-2) in response to this agent.21 The numbers of both T cells and NK cells are increased in patients receiving lenalidomide therapy22 ; however, NK-cell killing is also enhanced, including antibody-dependent cellular cytotoxicity and natural cytotoxicity.23,24 Moreover, these events correlate with clinical responses to lenalidomide therapy in patients with MM.22
In this report, we show that the PD-1/PD-L1 signaling axis mediates NK-cell activation and cytotoxicity against MM. We show that freshly isolated NK cells from healthy donors do not express PD-1; however, consistent with previous effects in T cells,25 exogenous IL-2 up-regulates its expression on NK cells. In contrast, PD-1 is expressed on freshly isolated NK cells from patients with active MM. CT-011 increases migration of NK cells toward MM targets mediated via the CXCR4/stromal-derived factor-1α (SDF-1α) pathway and enhances immune complex formation between patient-derived NK cells and PD-L1–bearing, primary autologous MM tumor cells. Moreover, lenalidomide down-regulates surface expression of PD-L1 on MM tumor cells. These events culminate in CT-011–mediated enhancement of patient-derived NK-cell activation and cytotoxicity specifically against autologous MM tumor cells but not against normal, healthy cells. Taken together, these findings provide novel mechanistic data and expand on previous translational and clinical experience with CT-011 to support the pursuit of a phase 2 clinical trial of CT-011 in combination with lenalidomide for patients with MM.
Methods
Primary cells and cell lines
All cells were cultured in RPMI 1640 media (Invitrogen) supplemented with 10% fetal bovine albumin (FBS; ICN Biomedicals) and kept at 37°C in 5% CO2. Primary human NK cells were isolated from leukopacks (American Red Cross) per an Ohio State University institutional review board–approved protocol as previously described and cultured in RPMI 1640 media with 10% FBS and 150 IU/mL IL-2 (Chiron).26 Peripheral blood mononuclear cells (PBMCs) and bone marrow aspirates from patients with MM were obtained under an institutional review board–approved procurement protocol. CD138+ cells were separated from whole marrow aspirates by the use of the EasySep Human Whole Blood CD138 Selection Kit (StemCell Technologies) per the manufacturer's instructions. The MM cell lines U266 and RPMI8226 as well as the NK cell–sensitive K562 cell line were obtained from ATCC and cultured in RPMI 1640 with 10% FBS.
Antibodies and reagents
CT-011 was kindly provided by CureTech, Ltd; all experiments described herein were conducted with a standard dose of 10 μg/mL. A phycoerythrin (PE)-labeled CT-011 was generated by Gerard Lozanski (The Ohio State University). Lenalidomide was kindly provided by John Byrd (The Ohio State University), and all experiments performed in the present work were conducted with a dose of 5nM.27 Anti–CD56-allophycocyanin (APC; no. IM2474U) was purchased from Beckman Coulter. Anti–CD3-PE-Cy7 (no. 557851) and anti–CD38-APC (no. 555462) were obtained from BD Pharmingen. Anti–CD138-fluorescein isothiocyanate (FITC; no. 11-1389-73), anti–CXCR4-APC (no. 17-9999-73), anti–PD-1-FITC (no. 11-9969-73), anti–CCR5-PE (no. 555993), anti-CXCR1-FITC (555939), anti–CXCR3-PE (no. 12-1831-82), anti-CCR7-FITC (no. 11-1979-73), and anti–PD-L1-PE (no. 12-5983-73) were purchased from eBioscience. Anti–CX3CR1-PE (no. D070-5) was obtained from MBL Corporation. Isotype controls were obtained from BD Biosciences. AMD-3100 (A5602-5MG) was purchased from Sigma-Aldrich.
Flow cytometric assays
Modulation of PD-L1 by lenalidomide was examined in the RPMI8226 MM cell line by recording mean fluorescent intensity (MFI) of the antigen at baseline and after incubation in lenalidomide or vehicle for 24 hours. Similarly, CD38+CD138+ malignant plasma cells were identified in whole marrow aspirates of patients, and in this gate, expression of PD-L1 was measured before and after 24 hours' incubation in lenalidomide or vehicle. The MFI of PD-L1 on CD38+CD138+ cells expressing PD-L1 was determined at baseline and after treatment. The MFIs of trafficking antigens were systematically evaluated on human NK cells at baseline and after stimulation with lenalidomide and/or CT-011 on an LSRII flow cytometer (BD Biosciences) and analyzed with FlowJo software (Version 8.8.6; Tree Star Inc).
Immune complex formation among primary, human effector NK cells, and primary MM tumor cell targets was examined with a 2-color staining and flow cytometric technique.28 Effector cells were labeled with carboxyfluorescein succinimidyl ester (CFSE; Sigma-Aldrich), and target cells were labeled with PKH dye (Sigma-Aldrich). NK cells were isolated as previously described and treated for 72 hours with CT-011 or control. After staining, 2.5 × 105 effectors and 2.5 × 105 target cells were pipetted into prechilled tubes on ice in the dark. To start the formation of immune complexes, the tubes were placed in a 37°C water bath for 2 minutes. When the incubation was complete, tubes were immediately removed, placed on ice, and ice-cold RPMI 1640 was added to stop the reaction. Data were collected by the use of a FACSCalibur flow cytometer (BD Biosciences) and analyzed with CellQuest software (BD Biosciences). Under control and experimental conditions, 10 000 total events were collected, and the total number of events in the right top quadrant (events double positive for PKH and CFSE) were studied. Stained target cells alone and stained effector cells alone were analyzed as controls.
A flow cytometric, target-based cytotoxicity assay was conducted according to methods previously published.29 NK cells were isolated as described previously and incubated for 72 hours in either control conditions or with lenalidomide, CT-011, or CT-011 and lenalidomide. Effectors were labeled with 3μM CFSE (from a 1mM stock solution in dimethyl sulfoxide) in phosphate-buffered saline (PBS) for 15 minutes at 37°C in a volume of 1 mL and then washed twice with PBS. Labeled NK cells were immediately mixed with a constant number of RPMI8226 targets at E:T ratios as described. In parallel, RPMI8226 cells were incubated alone to measure basal apoptosis. Cells were incubated for 4 hours at 37°C in a total volume of 150 μL of complete medium. Cell mixtures were then washed in PBS-1% FBS with 1% sodium azide and incubated with 10 μL of 7-amino-actinomycin D (BD Pharmigen) for 20 minutes at 4°C in the dark. After staining, cell mixtures were washed again with PBS-1% FBS with sodium azide and fixed with a 1% formalin solution. Data were collected on an LSRII flow cytometer (BD Biosciences) and analyzed with FlowJo software (Tree Star Inc).
Migration assay
Migration assays were performed with 24-well 6.5-mm Transwell plates and a 3.0μm Pore Polycarbonate Membrane Insert (Corning). Primary, human NK cells were prepared as described previously. The lower wells were filled in duplicate with 500 μL of either normal media (as a control for spontaneous migration), media from the U266 MM cell line, or serum from patients with MM. The polycarbonate membrane inserts were then placed in the upper chamber with 1 × 106 NK cells, pretreated as described previously, in 100 μL of normal media. After 4 hours at 37°C, the membrane inserts were removed, and the NK cells that had migrated to the bottom of each well were counted by the use of a Z1 Coulter Cell and Particle Counter (Beckman Coulter) and by light microscopy with a hemocytometer by trypan blue exclusion. Migration was determined by the following formula:
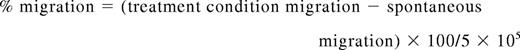
Elispot cytotoxicity assays
To measure NK-cell interferon-γ (IFN-γ) and granzyme B (GrB) secretion, enzyme-linked immunosorbent spot (ELISPOT) experiments were conducted with MultiScreen 96-well plates (Millipore) as described previously.30 Spots were visualized and counted with the Immunospot Imaging Analyzer (Cellular Technology Ltd).
Statistical analysis
We used t tests to compare both the percentage of cells positive for antigens of interest as well as the mean relative fluorescence intensity (MRFI) by flow cytometry. Analysis of variance (ANOVA) was used to determine statistical significance in migration assays, immune complex assays, and cytotoxicity assays. Planned comparisons for all ANOVA analyses were conducted by the method of Bonferroni. Log-transformation was applied to data of primary cell autologous cytotoxicity assays to stabilize variance. An unpaired Student t test was used to compare 2 independent groups, and a paired t test was used to compare 2 correlated groups, eg, the observations from the same donor. ANOVA was used when 3 or more independent groups were compared. To account for the correlation among replicates from the same patient, such as the immune complex assays and cytotoxicity assays, a mixed model was applied. The interaction contrast was applied to test the interaction between antibody treatment (PD-1 or control) and cell type (normal or tumor cell) for immune complex formation. For the directional hypothesis testing such as immune complex analysis, the one-sided test was used. Bonferroni method or the Holm step-down procedure was used to adjust P values for multiple comparisons. For other single comparisons, a P value less than .05 was considered statistically significant.
Results
PD-1 is absent on healthy donor NK cells but present on NK cells from patients with MM
Freshly isolated, resting, primary human CD56+CD3− NK cells from healthy donors do not constitutively express PD-1 (n = 5). However, PD-1 expression was inducible on healthy donor NKcells in response to 48 hours' IL-2 (150 IU/mL) stimulation as shown in a representative donor in Figure 1A (left). At baseline, whereas only 1.4% (± 0.35%, SD) of healthy donor NK cells expressed PD-1, 16% (± 6%) expressed PD-1 after 48 hours' IL-2 stimulation (representative example in the left panel of Figure1A; P = .001). The right panel of Figure 1A shows the MRFI of cells before (mean 1.06; ± 0.08 SD) and after the administration of IL-2 (4.25 ± 1.9; P = .02). Moreover, 64% (± 4.4%) of freshly isolated NK cells from patients with MM (n = 5) expressed PD-1 (P < .001). The average MFI of PD-1 on freshly isolated NK cells from patients with MM was 205 (± 59; representative example in left panel of Figure1B). The right panel of Figure1B shows the MRFI of healthy freshly isolated donor NK cells (1.02 ± 0.09) and patient-derived NK cells (3.07 ± 0.89; P = .006).
![Figure 1. PD-1 is differentially expressed on primary human NK cells as a function of MM disease activity. (A) By flow cytometry, PD-1 expression was measured in CD56+CD3− NK cells isolated from healthy donors (n = 5 donors). At baseline, virtually no expression was observed (1.4% ± 0.35% [SD] of NK cells per healthy donor sample); however, after IL-2 (150 IU/mL) stimulation for 48 hours, PD-1 expression became evident on an average of 16% (± 6%) of healthy donor NK cells, P = .001, a 12-fold increase (± 5-fold) in PD-1 expression. A representative sample is shown on left, and MRFI (mean ± SD) of pre–IL-2 baseline and post–IL2 PD-1 expression is shown on right. (B) In contrast to healthy donors, PD-1 is observed on freshly isolated NK cells from patients with MM (n = 5), a representative example is shown on left and MRFI (mean ± SD) of healthy donors and freshly isolated patient NK cells on right.](/view-large/figure/7104549/zh89991055700001.jpeg)
PD-1 is differentially expressed on primary human NK cells as a function of MM disease activity. (A) By flow cytometry, PD-1 expression was measured in CD56+CD3− NK cells isolated from healthy donors (n = 5 donors). At baseline, virtually no expression was observed (1.4% ± 0.35% [SD] of NK cells per healthy donor sample); however, after IL-2 (150 IU/mL) stimulation for 48 hours, PD-1 expression became evident on an average of 16% (± 6%) of healthy donor NK cells, P = .001, a 12-fold increase (± 5-fold) in PD-1 expression. A representative sample is shown on left, and MRFI (mean ± SD) of pre–IL-2 baseline and post–IL2 PD-1 expression is shown on right. (B) In contrast to healthy donors, PD-1 is observed on freshly isolated NK cells from patients with MM (n = 5), a representative example is shown on left and MRFI (mean ± SD) of healthy donors and freshly isolated patient NK cells on right.
PD-1 is differentially expressed on primary human NK cells as a function of MM disease activity. (A) By flow cytometry, PD-1 expression was measured in CD56+CD3− NK cells isolated from healthy donors (n = 5 donors). At baseline, virtually no expression was observed (1.4% ± 0.35% [SD] of NK cells per healthy donor sample); however, after IL-2 (150 IU/mL) stimulation for 48 hours, PD-1 expression became evident on an average of 16% (± 6%) of healthy donor NK cells, P = .001, a 12-fold increase (± 5-fold) in PD-1 expression. A representative sample is shown on left, and MRFI (mean ± SD) of pre–IL-2 baseline and post–IL2 PD-1 expression is shown on right. (B) In contrast to healthy donors, PD-1 is observed on freshly isolated NK cells from patients with MM (n = 5), a representative example is shown on left and MRFI (mean ± SD) of healthy donors and freshly isolated patient NK cells on right.
In addition, we confirmed previous work demonstrating PD-1 to be absent on freshly isolated CD3+ T cells from healthy donors, whereas 91% of freshly isolated CD3+ T cells from patients with MM expressed PD-1 (MFI = 652 ± 497, data not shown).
Low-dose lenalidomide down-regulates expression of PD-L1 on MM cell lines and primary MM tumor cells
The MM cell line RPMI8226 expresses PD-L1 (MFI = 174 ± 30 in 3 independent experiments). After 24 hours' incubation in lenalidomide, PD-L1 expression was significantly down-regulated (MFI = 47 ± 35; P = .01), as represented in top panel of Figure 2A (representative result of 3 independent experiments). The bottom panel of Figure 2A shows control and lenalidomide-treated MRFI of PD-L1 on the MM cell line (P = .04). We also confirmed work by previous groups that PD-L1 is selectively expressed on CD38+CD138+ malignant plasma cells in whole marrow aspirates from patients with MM (Figure 2B). Again, after a 24-hour incubation in low-dose lenalidomide (compared with incubation in medium alone), a 4-fold (± 1.9) reduction in PD-L1 expression was observed in CD138+CD38+ primary MM tumor cells (Figure 2C) as well (P < .05). (The top panel of Figure 2C is the representative result of 3 independent MM patient marrow samples. The bottom panel of Figure 2C shows control-treated and lenalidomide-treated MRFI of PD-L1 in primary MM tumor cells; P = .04.) Importantly, by MTS assay and trypan blue exclusion, in neither the lenalidomide-resistant RPMI8226 MM cell line nor the primary MM tumor cells was a direct effect on viability observed from this low dose of lenalidomide (data not shown).

Low-dose lenalidomide modulates PD-L1 expression on MM tumor cells. (A) The MM cell line RPMI8226 expresses high levels of PD-L1; however, after culture for 24 hours in lenalidomide (5nM), expression was markedly reduced (baseline, MFI 174 ± 30 vs lenalidomide-treated MFI, 47 ± 35; P = .01) without effect on cell viability (n = 3 independent experiments, top panel is representative result, bottom panel is MRFI of PD-L1 expression at baseline and in lenalidomide-treated cells). (B) We confirm previous observations that PD-L1 is uniquely expressed on CD38+CD138+ primary malignant plasma cells in whole-marrow aspirates (representative finding from n = 3 MM patients). (C) As in cell line experiments, lenalidomide (5nM) down-regulates PD-L1 expression without affecting cell viability (top panel is representative finding from n = 3 MM patients, bottom panel is MRFI of PD-L1 on control- and lenalidomide-treated cells, and dotted lines are isotype controls).
Low-dose lenalidomide modulates PD-L1 expression on MM tumor cells. (A) The MM cell line RPMI8226 expresses high levels of PD-L1; however, after culture for 24 hours in lenalidomide (5nM), expression was markedly reduced (baseline, MFI 174 ± 30 vs lenalidomide-treated MFI, 47 ± 35; P = .01) without effect on cell viability (n = 3 independent experiments, top panel is representative result, bottom panel is MRFI of PD-L1 expression at baseline and in lenalidomide-treated cells). (B) We confirm previous observations that PD-L1 is uniquely expressed on CD38+CD138+ primary malignant plasma cells in whole-marrow aspirates (representative finding from n = 3 MM patients). (C) As in cell line experiments, lenalidomide (5nM) down-regulates PD-L1 expression without affecting cell viability (top panel is representative finding from n = 3 MM patients, bottom panel is MRFI of PD-L1 on control- and lenalidomide-treated cells, and dotted lines are isotype controls).
CT-011 enhances NK-cell migration toward MM via the CXCR4/SDF-1α axis
We observed a morphologic change in the appearance of NK cells from healthy donors incubated with CT-011 (representative example at 40× magnification shown in supplemental Figure 1, available on the Blood Web site; see the Supplemental Materials link at the top of the online article), whereas there was no effect on proliferation or viability as was observed by MTS assay or manual counting by trypan blue exclusion. This finding led us to investigate the impact of CT-011 on NK-cell migration. We began by testing the effect of CT-011 on NK-cell trafficking toward MM cell line media and serum from a MM patient, each compared with normal medium, in a standard Transwell migration assay. As shown in Figure 3A, although CT-011 did not alter trafficking of NK cells into normal medium (white bars), significant increases in NK-cell trafficking were observed when NK cells were pretreated with CT-011 into medium from the U266 MM cell line (*P < .05 against control, grey bars) or serum from patients with MM (**P < .05 against control, black bars).

CT-011 and lenalidomide enhance NK-cell trafficking toward MM cell line media and MM patient serum. (A) To test whether or not CT-011 affects NK-cell trafficking, Transwell migration assays were conducted whereby NK cells were cultured in RPMI 1640 media with 10% FBS and 150 IU/mL IL-2 with isotype control or CT-011 for 72 hours. Data shown summarize 3 independent experiments. All differences within treatment conditions are statistically significant. Across treatment conditions, CT-011 enhanced migration beyond control into U266 media (*P < .05) and MM patient serum (**P < .05). (B) By flow cytometry, we systematically evaluated for changes in the surface expression of trafficking antigens known to be expressed by human NK cells. Only CXCR4 showed a statistically significant increase in expression in response to 72 hours in 150 IU/mL IL-2 with CT-011, P < .01 (representative data from one patient shown, n = 5). (C) Transwell migration assays were conducted with NK cells pretreated for 72 hours in 150 IU/mL IL-2 with control or CT-011 into normal media or media enriched with SDF-1α. CT-011 enhanced NK-cell trafficking into SDF-1α–enriched media ( , data collated from 3 independent experiments), suggesting a functional relevance to the increase in CXCR4 expression observed on NK cells in response to these agents. CT-011 enhanced migration over control condition (*P < .05). However, when NK cells were pretreated with the CXCR4 inhibitor AMD-3100 (10 μg/mL) for 90 minutes before assay, trafficking was virtually abolished in all conditions (
, data collated from 3 independent experiments), suggesting a functional relevance to the increase in CXCR4 expression observed on NK cells in response to these agents. CT-011 enhanced migration over control condition (*P < .05). However, when NK cells were pretreated with the CXCR4 inhibitor AMD-3100 (10 μg/mL) for 90 minutes before assay, trafficking was virtually abolished in all conditions ( , data from 2 independent experiments, pairwise comparisons within treatments; P < .05).
, data from 2 independent experiments, pairwise comparisons within treatments; P < .05).
CT-011 and lenalidomide enhance NK-cell trafficking toward MM cell line media and MM patient serum. (A) To test whether or not CT-011 affects NK-cell trafficking, Transwell migration assays were conducted whereby NK cells were cultured in RPMI 1640 media with 10% FBS and 150 IU/mL IL-2 with isotype control or CT-011 for 72 hours. Data shown summarize 3 independent experiments. All differences within treatment conditions are statistically significant. Across treatment conditions, CT-011 enhanced migration beyond control into U266 media (*P < .05) and MM patient serum (**P < .05). (B) By flow cytometry, we systematically evaluated for changes in the surface expression of trafficking antigens known to be expressed by human NK cells. Only CXCR4 showed a statistically significant increase in expression in response to 72 hours in 150 IU/mL IL-2 with CT-011, P < .01 (representative data from one patient shown, n = 5). (C) Transwell migration assays were conducted with NK cells pretreated for 72 hours in 150 IU/mL IL-2 with control or CT-011 into normal media or media enriched with SDF-1α. CT-011 enhanced NK-cell trafficking into SDF-1α–enriched media (, data collated from 3 independent experiments), suggesting a functional relevance to the increase in CXCR4 expression observed on NK cells in response to these agents. CT-011 enhanced migration over control condition (*P < .05). However, when NK cells were pretreated with the CXCR4 inhibitor AMD-3100 (10 μg/mL) for 90 minutes before assay, trafficking was virtually abolished in all conditions (, data from 2 independent experiments, pairwise comparisons within treatments; P < .05).
We systematically evaluated the effects of CT-011 on the expression of a panel of trafficking antigens expressed on human NK cells (n = 5), including CXCR4, CCR5, CCR7, CX3CR1, CXCR1, and CXCR3.31 Among all antigens examined, only a modest, but statistically significant increase in expression of CXCR4 was observed in response to CT-011 on NK cells, baseline MFI = 789 (± 272) versus CT-011-treated (924 ± 262, P < .01; a representative example of n = 5 is shown in Figure 3B).
To test the functional consequence of CXCR4 up-regulation by CT-011, we evaluated the effects of CT-011 on NK-cell trafficking toward SDF-1α (1 ng/mL)–enriched media compared with normal media. As before, CT-011 did not enhance NK-cell trafficking into normal media; however, NK cells pretreated with CT-011 showed enhanced migration into SDF-1α–enriched media (Figure 3C light gray bars; *P = .001). Furthermore, when NK cells were pretreated with CT-011 but then exposed to the CXCR4 antagonist AMD-3100 (10 μg/mL) for 90 minutes before the experiment, migration was essentially abolished (Figure 3C dark gray bars, data from 2 independent experiments).
CT-011 enhances immune complex formation between NK cells and PD-L1–bearing tumor targets
By using a previously validated flow cytometric technique to assess immune complex formation between effector and target cells,28 we studied the effects of CT-011 on primary, human NK cells versus PD-L1–bearing tumor cell immune complex formation. Primary human NK cells labeled with CFSE were treated with CT-011 or control and were cocultured with primary human MM labeled with PKH dye. Data were collected regarding CFSE-positive, PKH-positive, and double-positive events, the latter interpreted as immune complexes between NK-cell effectors and tumor cell targets. In control conditions, few double-positive events were observed. The left panel of Figure 4A shows a representative result of immune complex formation in control conditions. As shown in the right panel of Figure 4A, a representative increase in immune complex formation between CT-011–treated NK-cell effectors and MM tumor targets was observed. Figure 4B shows data from immune complex experiments among primary human NK cells and the NK-cell–sensitive K562 cell line, the MM cell lines U266 and RPMI8226, and primary human MM tumor cells. (We verified that K562 expresses PD-L1 by flow cytometry; data not shown.) NK cells pretreated with CT-011 showed a 2.2-fold (± 0.22-fold, SEM) increase in immune complex formation against K562 (Figure 4A; *P = .02), a 2.3-fold (± 0.14-fold) increase against U266 (Figure 4A; **P = .007), a 1.55-fold (± 0.06-fold) increase against RPMI8226 (Figure 4A; ***P = .01), and a 1.4-fold (± 0.06-fold) increase against primary, human MM cells (Figure 4A; ****P = .009).

CT-011 enhances immune complex formation between effector NK cells and target MM cells. (A) A representative example of the flow cytometric technique to evaluate immune complex formation is shown, where primary, human NK cells, cultured as before for 72 hours in IL-2 and control mAb or CT-011, were stained with CFSE. Primary MM tumor cell targets were isolated from MM patient marrow aspirates and stained with PKH. Flow gates were created with PKH+ events representing MM target cells on the y-axis and CFSE+ effector NK cells on the x-axis. Double-positive events (ie, PKH+CFSE+) were interpreted as immune complex formations (shaded right top panel of flow diagrams). The left panel is representative of findings in control conditions; the right panel is representative of findings where NK cells were pretreated with CT-011. (B) Immune complex formation between NK cells and the K562, U266, RPMI8226 cell lines as well as with primary MM tumor cells was evaluated. At baseline, less than 1% of gated events in all conditions were PKH+CFSE+. However, NK cells pretreated with CT-011 led to statistically significant increases in immune complex formation with K562 (*P = .02), U266 (**P = .007), RPMI8226 (***P = .01), and primary MM tumor cell targets (****P = .009). All results shown are from at least 3 independent experiments with each target. (C) In this figure, total PBMCs obtained from patients with MM (rather than purified NK cells as in panels A-B) were treated for 48 hours in IL-2 with control or CT-011 mAb and immune complex formation in response to CT-011 against autologous CD138+PD-L1+ MM tumor cells or CD138−PD-L1− cellular marrow elements was compared. Compared with control-treated effector cells, CT-011 increased immune complex formation with MM tumor cells (*P < .001) but not against normal marrow cells. Data shown are from 3 independent experiments in n = 3 patients.
CT-011 enhances immune complex formation between effector NK cells and target MM cells. (A) A representative example of the flow cytometric technique to evaluate immune complex formation is shown, where primary, human NK cells, cultured as before for 72 hours in IL-2 and control mAb or CT-011, were stained with CFSE. Primary MM tumor cell targets were isolated from MM patient marrow aspirates and stained with PKH. Flow gates were created with PKH+ events representing MM target cells on the y-axis and CFSE+ effector NK cells on the x-axis. Double-positive events (ie, PKH+CFSE+) were interpreted as immune complex formations (shaded right top panel of flow diagrams). The left panel is representative of findings in control conditions; the right panel is representative of findings where NK cells were pretreated with CT-011. (B) Immune complex formation between NK cells and the K562, U266, RPMI8226 cell lines as well as with primary MM tumor cells was evaluated. At baseline, less than 1% of gated events in all conditions were PKH+CFSE+. However, NK cells pretreated with CT-011 led to statistically significant increases in immune complex formation with K562 (*P = .02), U266 (**P = .007), RPMI8226 (***P = .01), and primary MM tumor cell targets (****P = .009). All results shown are from at least 3 independent experiments with each target. (C) In this figure, total PBMCs obtained from patients with MM (rather than purified NK cells as in panels A-B) were treated for 48 hours in IL-2 with control or CT-011 mAb and immune complex formation in response to CT-011 against autologous CD138+PD-L1+ MM tumor cells or CD138−PD-L1− cellular marrow elements was compared. Compared with control-treated effector cells, CT-011 increased immune complex formation with MM tumor cells (*P < .001) but not against normal marrow cells. Data shown are from 3 independent experiments in n = 3 patients.
Given that the latter findings were performed with healthy donor NK cells against allogeneic primary MM tumor targets, we sought to validate these observations by using patient-derived effector cells against autologous MM tumor targets. To determine the specificity of this effect, we evaluated the effects of CT-011 on the ability of patient-derived PBMC effector cells to form immune complexes with both autologous CD138+PD-L1+ MM tumor cells and CD138−PD-L1− marrow cells (under these circumstances, we were unable to enrich for sufficient numbers of NK cells). PBMCs were isolated from patients with MM (n = 3), and CD138+PD-L1+ MM tumor cells were isolated from whole marrow aspirates. Effector cells were pretreated for 72 hours with CT-011 alone or in control conditions and immune complex formation against autologous CD138+PD-L1+ MM tumor cells was compared with immune complex formation against CD138−PD-L1− cellular marrow elements. Immune complex formation observed in CT-011–treated effectors against MM target cells and normal marrow cells is shown in Figure 4C. The interaction between MM targets and normal marrow elements was significant (P < .001). CT-011–treated PBMCs formed 1163 (± 49 SEM) immune complexes with autologous MM cells compared with 633 (± 73) in control-treated PBMCs against autologous MM (Figure 4C left; *P < .001). No differences were observed in the rate of immune complex formation with normal marrow elements in response to CT-011 compared with control-treated PBMC (Figure 4C right; P = .547).
CT-011 enhances NK-cell GrB and IFN-γ production against PD-L1–bearing tumor targets
By using an effector-based ELISPOT assay, we studied the effects of CT-011 on primary, human NK-cell production of GrB against the K562 cell line and RPMI8226 MM cell line (E:T = 10:1). After more than 3 independent experiments (Figure 5A), CT-011 led to a statistically significant increase in GrB production against K562 (Figure 5A left; *P < .05) and RPMI8226 (Figure 5A right; **P < .05). In a similar ELISPOT assay (Figure 5B), CT-011 also increased NK-cell IFN-γ secretion against primary MM cell targets. Control-treated NK cells produced an average of 483 (± 14) spots/well, whereas CT-011–treated NK cells produced an average of 708 (± 43) spots/well of IFN-γ, (Figure 5B; *P < .001).

CT-011 enhances NK-cell GrB and IFN-γ production against PD-L1–bearing tumor cell targets. (A) By using an ELISPOT effector-based assay, we studied the effects of CT-011 on GrB production by primary, human NK cells against the K562 and RPMI8226 cell lines (E:T = 10:1). After 72 hours of pretreatment with 150 IU/mL IL-2 and control or CT-011 mAb, the latter led to statistically significant increases in NK-cell degranulation of GrB against K562 (*P < .05) and RPMI8226 (**P < .05; data shown are representative of 3 independent experiments against each line). (B) By using the same effector-based ELISPOT cytotoxicity assay, we found that CT-011 also enhanced IFN-γ production by NK cells (pretreated for 72 hours in IL-2 150 IU/mL and control or CT-011 mAb) against primary MM tumor cell targets (*P = .01; data shown from 3 independent experiments). (C) By using a target-based cytotoxicity assay, primary, we pretreated human NK cells for 72 hours in IL-2 150 IU/mL and control, lenalidomide (5nM), CT-011, or the combination. Lenalidomide and CT-011 statistically significantly increased cytotoxicity against RPMI8226 cell line targets (E:T 50:1, *P = .03, ET:100:1, **P = .02; data shown are from 3 independent experiments at both E:T ratios). (D) PBMCs and marrow aspirates were obtained from patients with MM (n = 3) and CD138+ tumor cells were isolated from the whole marrow aspirate. Effector cells were cultured in IL-2 and control or CT-011 for 48 hours and CD138+PD-L1+ MM tumor cells and CD138−PD-L1− cellular marrow fraction served as target populations. On the left, cytotoxicity is enhanced against autologous CD138+PD-L1+ MM tumor cells (*P = .005; data shown from n = 3 independent experiments), yet no increase in cytotoxicity is conferred against autologous CD138−PD-L1− cellular marrow elements (right; P = ns).
CT-011 enhances NK-cell GrB and IFN-γ production against PD-L1–bearing tumor cell targets. (A) By using an ELISPOT effector-based assay, we studied the effects of CT-011 on GrB production by primary, human NK cells against the K562 and RPMI8226 cell lines (E:T = 10:1). After 72 hours of pretreatment with 150 IU/mL IL-2 and control or CT-011 mAb, the latter led to statistically significant increases in NK-cell degranulation of GrB against K562 (*P < .05) and RPMI8226 (**P < .05; data shown are representative of 3 independent experiments against each line). (B) By using the same effector-based ELISPOT cytotoxicity assay, we found that CT-011 also enhanced IFN-γ production by NK cells (pretreated for 72 hours in IL-2 150 IU/mL and control or CT-011 mAb) against primary MM tumor cell targets (*P = .01; data shown from 3 independent experiments). (C) By using a target-based cytotoxicity assay, primary, we pretreated human NK cells for 72 hours in IL-2 150 IU/mL and control, lenalidomide (5nM), CT-011, or the combination. Lenalidomide and CT-011 statistically significantly increased cytotoxicity against RPMI8226 cell line targets (E:T 50:1, *P = .03, ET:100:1, **P = .02; data shown are from 3 independent experiments at both E:T ratios). (D) PBMCs and marrow aspirates were obtained from patients with MM (n = 3) and CD138+ tumor cells were isolated from the whole marrow aspirate. Effector cells were cultured in IL-2 and control or CT-011 for 48 hours and CD138+PD-L1+ MM tumor cells and CD138−PD-L1− cellular marrow fraction served as target populations. On the left, cytotoxicity is enhanced against autologous CD138+PD-L1+ MM tumor cells (*P = .005; data shown from n = 3 independent experiments), yet no increase in cytotoxicity is conferred against autologous CD138−PD-L1− cellular marrow elements (right; P = ns).
CT-011 alone or with lenalidomide enhances NK-cell cytotoxicity against MM tumor cell targets
In a complementary manner to the effector-based assays shown previously, we validated these results by using a target-based flow cytometric cytotoxicity technique.29 In identical effector treatment conditions, the combination of lenalidomide and CT-011 (Figure 5D black bar) significantly enhanced NK-cell cytotoxicity versus control (Figure 5D white bar) against RPMI8226 MM cell line targets. Results shown in Figure 5C are from 3 independent experiments at E:T 50:1 (left; P = .03) and 100:1 (right; P = .02). Similar results were obtained by the use of the MM cell line U266 as targets in 2 additional experiments (data not shown). Interestingly, in contrast to these results, a brief 30-minute incubation of NK cells with CT-011 before coculture with MM targets did not enhance cytotoxicity, suggesting that additional mechanisms beyond merely preventing receptor/ligand interaction may be involved in the enhanced cytotoxicity observed with CT-011 (data not shown). In addition, we also repeated this cytotoxicity assay with an anti-CD56 IgG1 antibody (as an additional control condition to use an antibody that would directly react with, but not activate, NK cells), and in this instance, incubation with the anti-CD56 control led to similar results as the nonspecific IgG1 isotype (data not shown).
To confirm these findings in an autologous setting, we procured PBMCs and whole marrow aspirates from patients with MM (n = 3). PBMCs were cultured in control conditions or with CT-011 for 72 hours. CD138+PD-L1+ MM tumor cells were isolated from whole marrow aspirates by a passive selection technique; purity was greater than 90% by flow cytometry (data not shown). Target-based cytotoxicity assays identical to those presented previously were conducted by use of CD138+PD-L1+ MM tumor cells as targets in one condition and the CD138−PD-L1− cellular marrow fraction as targets in the other condition. As shown in Figure 5D (E:T 20:1), significant differences were observed in cytotoxicity as a function of target-cell population (P < .001). Pretreatment of patient effector cells with CT-011 (dark bars) specifically enhanced cytotoxicity against the autologous CD138+PD-L1+ MM tumor cells (72% ± 9%) compared with pretreatment of effector cells in control conditions (29% ± 5%; P = .005; Figure 5D left). Importantly, no increase in cytotoxicity was observed against CD138−PD-L1− normal cellular marrow elements (2.8% ± 0.13% for CT-011 pretreated effector cells vs 2.6% ± 0.2% for control; P = .5; Figure 5D right).
Discussion
The PD-1/PD-L1 axis has been implicated in tumor immunoevasion of the T-cell–mediated, adaptive immune response in many human solid tumors32-35 as well as several B-cell hematologic malignancies, including Hodgkin lymphoma, non-Hodgkin lymphoma, and MM.18,19,36 Until now, to our knowledge, little research has characterized the role of the PD-1/PD-L1 axis in regard to NK cell–mediated innate immune response to MM or other tumors. Here we show that NK cells from patients with MM selectively express PD-1 and confirm that the MM cells from these patients selectively express its cognate ligand, PD-L1. We provide novel data to suggest that interruption of the PD-1/PD-L1 axis by CT-011 enhances primary, human NK cell–mediated function against autologous MM cells. Further we provide evidence for the mechanisms by which this may be occurring. Finally, we show that lenalidomide can down-modulate PD-L1 from the surface of primary MM cells, providing a novel mechanism by which this drug may be augmenting the NK cell versus MM effect.
NK cells are capable of recognizing and killing MM tumor cells1-4 ; however, this effect is attenuated with progression of disease through several mechanisms, including the up-regulation of ligands that inhibit the immune response, such as PD-L1.18,19 Recent work by our group and others, however, has shown the NK-cell versus MM effect can be recovered and augmented in a therapeutic manner.11,12,37 Several findings in the present work allow us to extend this idea further and identify the PD-1/PD-L1 signaling axis as a therapeutic target in patients with MM.
Here we provide new evidence that PD-1 is expressed on freshly isolated NK cells from patients with MM but not on human NK cells isolated from healthy donors. Coupled with the established finding that MM tumor cells express PD-L1,19 the present work highlights the PD-1 pathway as an important immunoevasive mechanism in MM. Our work adds to a growing list of strategies through which the malignant plasma cell clone may evade the NK-cell immune response, including up-regulation of other inhibitory ligands (eg, major histocompatibility complex class 1)38 and the shedding of soluble forms of activating ligands (eg, major histocompatibility complex class I–related chain A).39
Second, we show that a low dose of lenalidomide can down-regulate expression of PD-L1 on primary MM tumor cell targets independent of a direct apoptotic effect. Because lenalidomide also exerts anti-MM efficacy through expansion and activation of the NK-cell compartment through T-cell production of IL-2, we hypothesized that adding CT-011 to lenalidomide would further enhance the NK-cell versus MM effect.11
Third, we found that CT-011 enhances NK-cell migration toward malignant plasma cells. SDF-1α, the cognate ligand for CXCR4, is produced and expressed by bone marrow stromal cells and plays an important role in the development and progression of MM.40-42 Patients with MM have elevated levels of the cognate chemokine for CXCR4, SDF-1α.43 In addition, CXCR4 has been shown to mediate NK-cell trafficking to the bone marrow microenvironment.32,44 Thus, these findings may have in vivo relevance, ie, enhancing NK-cell trafficking to bone marrow in patients with MM. Moreover, the alteration in appearance in NK cells induced by CT-011 (supplemental Figure 1) as well as the increased expression of CXCR4 suggest that ligation of PD-1 by CT-011 alters NK-cell function beyond merely blocking interaction between PD-1 and associated ligands. The molecular mechanisms of these findings remain unclear; however, preliminary data and previous work suggest that phosphoinositide-3-kinase pathway signaling via the PD-1 intracellular immunoreceptor tyrosine-based switch motif may mediate effects on actin cytoskeletal remodeling and NK cell trafficking.45,46
As opposed to T and B cells, NK cells do not require gene rearrangement or costimulatory signals to initiate an immune response. Rather, once an NK cell acquires a potential target, the decision to kill or not depends on the balance between activating and inhibitory signals received through a complex array of receptors displayed on the NK cell.47 For instance, NK cells must express at least one of the activating receptors, DNAM-1, NKG2D, or NKp46, to exert cytotoxicity against MM targets.48 However, inhibitory receptor/ligand interaction and consequent signaling has been shown to prevent initiation of cytotoxicity, even in the presence of activating receptor/ligand interaction and signaling.47 Therefore, that pretreatment of NK cells with CT-011 alone or with lenalidomide enhances immune complex formation with MM tumor cell targets is an especially relevant finding. (Interestingly, the absolute numbers of immune complexes observed between patient-derived PBMCs and autologous MM targets [Figure 4C] are greater than those observed between purified NK cells and allogeneic targets [Figure 4B]. The reason for this difference is unclear but may be related to immune complexes between PD-1–bearing CD3+ T cells and MM targets in addition to NK cells and MM targets.) This finding directly supports the idea that the PD-1/PD-L1 signaling axis contributes to MM tumor cell immunoevasion of NK-cell surveillance and immunity. (The finding that CT-011 enhanced NK-cell function against the K562 cell line, which lacks MHC class 1 expression, is provocative as well, suggesting that the PD-1/PD-L1 axis may convey an additional and unique contribution to tumor cell immunoevasion beyond other established mechanisms.)
Moreover, these events culminate in an enhancement of NK-cell cytotoxicity with CT-011 and lenalidomide against MM tumor cells, as demonstrated through complementary effector- and target-based assays. Importantly, our results suggest that the increase in cytotoxicity conferred by CT-011 is specifically directed against the PD-L1–bearing, CD138+ malignant plasma cell clone in patients with MM because CD138−,PD-L1− normal cellular marrow elements were spared in autologous killing assays (Figure 5D). Notably, caution is required in ascribing the entirety of the improvement in tumor-specific cytotoxicity in the autologous killing assay (Figure 5D), given that CT-011 may also have affected PD-1–bearing CD3+ T cells as well in this setting.
These data build on previous work characterizing the enhancing effects of CT-011 on adaptive effector cell populations against tumor cells by extending these findings into the realm of innate immunity and MM.15-17 In addition, the present human data provide novel in vitro mechanisms by which CT-011 enhances innate immune response previously described in an in vivo model.16 In a phase 1 clinical trial of CT-011, a single intravenous dose led to expansion of T-cell subsets as well as single-agent, clinical benefit responses in 6 of 17 treated patients with advanced hematologic malignancies, including 1 complete response, 1 minor response, and 4 stable disease, although no correlative data were available on the NK-cell compartment of treated patients.20 Given the direct anti-MM effect of lenalidomide as well as the complementary mechanisms of action described herein with CT-011, a phase 2 trial of the combination appears justified in patients with MM. The agents share no predicted cross-toxicities, and the combination also is devoid of concomitant corticosteroid administration. CT-011 and lenalidomide may represent a promising dual immunotherapy for patients with MM.
Portions of this work were presented at the American Association for Cancer Research 100th Annual Meeting, Denver, CO, April 18-22, 2009.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was funded by American Cancer Society Grant no. IRG-67-003-44 (to D.M.B.) and funding from MMORE (Multiple Myeloma Opportunities for Research and Education; to D.M.B., C.C.H., Y.E.).
Authorship
Contribution: D.M.B. designed research, performed experiments, analyzed data, and wrote the manuscript; C.E.B. designed research, performed experiments and wrote the manuscript; A.M. designed and performed experiments; C.C.H., Y.E., M.K.S., and C.G. designed research, procured patient samples, and assisted in writing the manuscript; B.B., R.A.B., and J.Y. designed research, analyzed data, and assisted in writing the manuscript; G.L. and J.C.B. contributed vital reagents; P.P. and S.M.D. designed research; J.Z. performed statistical analyses and assisted in manuscript writing; R.R.Y. contributed vital reagents and assisted in writing the manuscript; and M.A.C. designed experiments and assisted in writing the manuscript.
Conflict-of-interest disclosure: R.R.Y. is an employee of CureTech Ltd, which provided CT-011. The remaining authors declare no competing financial interests.
Correspondence: Don M. Benson Jr, MD, PhD, A445-A Starling Loving Hall, 320 W 10th Ave, Columbus, OH 4321-1240; e-mail: Don.benson@osumc.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal