

Abstract
AML1-ETO is the chimeric protein product of the t(8;21) in acute myeloid leukemia. The ETO portion of the fusion protein includes the eTAFH domain, which is homologous to several TATA binding protein–associated factors (TAFs) and interacts with E proteins (E2A and HEB). It has been proposed that AML1-ETO–mediated silencing of E protein function might be important for t(8;21) leukemogenesis. Here, we determined the solution structure of a complex between the AML1-ETO eTAFH domain and an interacting peptide from HEB. On the basis of the structure, key residues in AML1-ETO for HEB association were mutated. These mutations do not impair the ability of AML1-ETO to enhance the clonogenic capacity of primary mouse bone marrow cells and do not eliminate its ability to repress proliferation or granulocyte differentiation. Therefore, the eTAFH-E protein interaction appears to contribute relatively little to the activity of AML1-ETO.
Introduction
AML1-ETO is the fusion protein produced from the t(8;21) identified in acute myeloid leukemia (AML) of the M2 subtype.1 The translocation fuses the N-terminus of RUNX1 with 575 amino acids of ETO (“eight twenty one,” encoded by RUNX1T1). AML1-ETO has 5 conserved domains named for its Drosophila homologues Runt and Nervy: the DNA- and CBFβ-binding Runt domain from RUNX1, and Nervy homology domains 1 to 4 (NHR1-4) in ETO.2-4 NHR1, also known (and referred to here) as the TATA box-binding protein-associated factor homology domain (eTAFH) is homologous to several TATA-binding protein associated factors (TAFs) and interacts with E proteins.5 The NHR2 domain, or hydrophobic heptad repeat (HHR) is an α-helical tetramer that is essential for the activity of AML1-ETO.6-9 NHR3 is a predominantly α-helical domain that interacts with the regulatory subunit of type II cyclic AMP-dependent protein kinase (PKA RIIα).4 The NHR4 domain (also known as myeloid-Nervy-DEAF-1, or MYND) was suggested to contain 2 putative non–DNA-binding zinc-fingers.10-12 We previously solved the structure of the MYND domain and showed it to be a member of the RING finger structural family.13
The eTAFH domain is homologous to several TAFs, including Drosophila TAF110, human TAF130, and human TAF105.3,14,15 Previous studies in a t(8;21) leukemic cell line showed that the eTAFH domain interacts with E proteins,5 which are widely expressed DNA-binding transcription factors involved in regulating differentiation, proliferation, and apoptosis whose transactivation is facilitated by recruiting CBP/p300 histone acetyltransferases (HATs).16-18 The eTAFH domain binds the AD1 transactivation domain of the HeLa cell E-box-binding protein (HEB), an E protein family member, which prevents CBP/p300 and HAT association. This led to the hypothesis that AML1-ETO dominantly silences E proteins in t(8;21) cells, resulting in dysregulation of E protein target genes, and by that mechanism contributes to leukemogenesis.5 Consistent with this, a recent chromatin immunoprecipitation (ChIP)–chip study shows evidence for localization of AML1-ETO and HEB on chromatin.19
Structural characterization of the interaction between the eTAFH domain and the E proteins is necessary to understand the basis for selective binding and to develop highly specific point mutations to study the role of this interaction in AML1-ETO leukemogenesis. Although the eTAFH domain structure was recently described by 2 different groups, the structure of a complex between the eTAFH domain and an E protein was not determined.20,21 Both groups reported docking models of a HEB AD1 domain with eTAFH, but our results show that these models are not accurate. Here, we report the structure of a TAFH domain–HEB peptide complex determined with solution nuclear magnetic resonance (NMR) spectroscopy. This structure yields a detailed understanding of the recognition elements for HEB binding to the eTAFH domain. Using the structure, we have identified point mutations that disrupt HEB binding while preserving the fold of the domain for subsequent functional studies. These mutations were introduced into AML1-ETO, and effects on the serial replating, differentiation, and proliferation of primary mouse bone marrow cells enriched for hematopoietic progenitors were evaluated. These mutations do not eliminate the ability of AML1-ETO to enhance the clonogenic capacity of primary mouse bone marrow cells, nor do they attenuate its activity in proliferation or in granulocyte differentiation.
Methods
Protein expression and purification
cDNA-encoding eTAFH residues G267 to Q364 of AML1-ETO (GenBank ID, 407727) were cloned between BamH1 and XhoI sites of a pHis-parallel vector.22 Mutations in eTAFH were introduced using the QuickChange Site Directed Mutagenesis kit (Stratagene, La Jolla, CA). Coding sequences for the HEB peptide and a 14-residue linker were ligated to the 3′ end of those encoding eTAFH by standard polymerase chain reaction (PCR) cloning procedures. The wild-type eTAFH, mutant eTAFHs, and eTAFH-HEB were expressed in Rosetta (DE3) cells by inducing with 0.8 mM IPTG at 30°C when OD600 nm reached 0.5 to 0.6. The cell lysate was applied to a Ni-NTA column (QIAGEN, Valencia, CA), the eluate was dialyzed against 2 mM Bis-Tris (pH 6.0), 50 mM NaCl, and 1 mM DTT to remove the imidazole, and then applied to Ni-NTA column after cleaving the 6 × His-tag with the protease AcTEV (Invitrogen, Carlsbad, CA). The nonadherent fraction was purified by ion-exchange chromatography on an SP-Sepharose column (GE Healthcare, Little Chalfont, United Kingdom).
Isothermal titration calorimetry
Isothermal titration calorimetry (ITC) experiments were carried out on a VP-ITC MicroCalorimeter system (MicroCal Inc, Northampton, MA) at 25°C. Protein samples were dialyzed against 25 mM Tris-HCl (pH 7.5), 50 mM NaCl, and 1 mM EDTA, centrifuged, and degassed before use. A solution of 58 μM wild-type eTAFH was titrated with 590 μM HEB peptide, and 64 μM L325E and 105 μM F332A were titrated with 740 μM and 550 μM HEB peptide, respectively. Data were corrected for dilution enthalpy and then analyzed using Origin 7.0 (Origin Lab, Northampton, MA).
NMR spectroscopy
All NMR experiments except NOESY spectra were performed at 30°C on a Varian Inova 500 MHz or 600 MHz (Palo Alto, CA) equipped with a cryogenic probe. 15N-NOESY-HSQC and 13C-NOESY-HSQC were acquired at 30°C on a Bruker Avance 900 MHz at the Nuclear Magnetic Resonance Facility (Madison, WI). All NMR samples were prepared in 25 mM Bis-Tris (pH 6.0), 350 mM NaCl, 1 mM EDTA, and 5% (vol/vol) D20. U-[15N, 13C]-labeled eTAFH-HEB was prepared for conventional triple resonance experiments. For the stereospecific assignments of methyl groups of leucines and valines, 10% fractional 13C-labeled eTAFH-HEB was prepared by growing cells in minimal media containing 90% 12C-glucose and 10% 13C-glucose. U-[15N, 13C, 2H]-labeled eTAFH-HEB was soaked into a 6% stretched polyacrylamide gel for collecting residual dipolar coupling data. Three types of RDCs, 1DHN, 1DNC′, and 1DC′Cα, were measured by HNCO-based experiments.23
Titration of eTAFH with peptide ligands monitored by NMR
1HN and 15N chemical shift changes in eTAFH were measured on titration of 100 μM U-15N-labeled eTAFH with each peptide by following peak movements in 15N-1H HSQC spectra. The Kd for binding of eTAFH and each peptide was determined by least-squares fitting of chemical shift changes as a function of ligand concentration according to the equation:
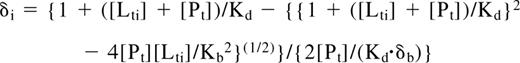
Structure calculation
We calculated the 3D structure of eTAFH-HEB with CNS using the simulated annealing protocol. NOE restraints were obtained from manual assignment and classified into 4 groups: 1.8 to 2.8, 1.8 to 3.3, 1.8 to 4.2, and 1.8 to 5.5 Å. The φ and ψ dihedral angle restraints were generated by TALOS (torsion angle likelihood obtained from shift and sequence similarity) prediction based on Cα, Cβ, C′, and N chemical shifts. Stereospecific assignments of methyl protons of leucines and valines were obtained from analyzing 13C-1H HSQC of fractional 13C-labeled protein.26 3JHNHA-coupling constant restraints were obtained from 3D HNHA spectra. Three sets of RDCs (1DHN, 1DNC′, and 1D C{prime]Cα) were used in the structure calculation. Initial values of the Da and R of the alignment tensor were estimated from a histogram of RDC distribution. The 20 lowest energy structures of 200 calculated structures were selected to present the structure.
Coimmunoprecipitations
Cos7 cells in 15-cm plates were transfected with 4.5 μg plasmid DNA expressing FLAG-tagged HEB and a microgram amount of plasmid DNA expressing equivalent levels of AML1/ETO and its mutants as determined by titration experiments using FUGENE6 (Roche, Indianapolis, IN) and cultured for 24 hours in DMEM supplemented with 10% fetal calf serum. HEB was immunoprecipitated using anti–FLAGM2-conjugated agarose beads following the manufacturer's instructions (Sigma, Poole, United Kingdom). Immunoprecipitates were washed once using 1% Triton X lysis buffer and twice in Tris-buffered saline (TBS), pH 8.0. Immune complexes were boiled in SDS buffer, and immunoprecipitates were analyzed by Western blot using anti-FLAG or monoclonal anti-Runt domain antibodies.
Retroviral transduction
Mutated AML1-ETO proteins were transferred from pBluescript into the MIGR1 vector,27 and retroviruses were prepared as described previously.6 Primary bone marrow mononuclear cells were harvested from 5- to 6-week-old C57BL/6 mice (The Jackson Laboratory, Bar Harbor, ME) and subjected to immunomagnetic-negative selection using the Lineage cell depletion Kit (Miltenyi Biotec, Auburn, CA). Cells (6 × 105 in 3 mL) were plated in ultralow adhesion 6-well plates (Costar, Corning, NY) and incubated overnight at 37°C, 5% CO2 in RPMI containing 20% FCS, penicillin and streptomycin, 10 ng/mL IL-3, 20 ng/mL IL-6 (R&D Systems, Minneapolis, MN), and 20 ng/mL SCF (StemCell Technologies, Vancouver, BC). Twelve hours later, 6 × 105 cells in 2 mL fresh media were added to 6-well plates (Cellstar; Greiner Bio-One, Monroe, NC) coated with 100 μg Retronectin (Takara, Madison, WI). Retroviral supernatants (2 mL), 4 μL of 40 mg/mL polybrene, and 40 μL of 1 M HEPES were added to each well, and the cells were centrifuged for 90 minutes at 1400g, 37°C. Animal work was approved by the Institutional Animal Care and Use Committee at Dartmouth (Hanover, NH).
Granulocyte differentiation
Granulocyte differentiation was assessed as described previously.6 Bone marrow (BM) was harvested from male C57BL/6 mice and cultured for 2 days in DMEM plus FCS P/S, 10 ng/mL IL-3, 20 ng/mL IL-6, 100 ng/mL SCF (R&D Systems). Cells expressing lineage markers were depleted using a cocktail of antibodies conjugated to magnetic beads (Miltenyi Biotec). Lineage-negative (Lin−) cells were infected with retroviruses expressing GFP alone, or GFP in combination with AML1-ETO proteins, and cultured for 7 days in DMEM FCS P/S, 10 ng/mL IL-3, 20 ng/mL IL-6, 100 ng/mL SCF, 60 ng/mL G-CSF (R&D Systems). Cells were harvested and stained for surface expression of Mac-1 and Gr-1 (Ly-6C) with antibodies conjugated to phycoerythrin (PE) or allophycocyanin (APC), respectively, with the use of a FACSCalibur flow cytometer (BD Bioscience, San Jose CA). The data were analyzed using FlowJo software (version 6.1.1; TreeStar, San Carlos, CA).
Serial replating
Immediately after retroviral transduction, 103 cells were plated in M3434 complete methylcellulose media (StemCell Technologies) and cultured for 7 days. After colonies were enumerated, the cultures were diluted, the cells were resuspended, and 1 × 104 cells were replated every 7 days for a total of 28 days.
Cell cycle kinetics
BrdU incorporation by Lin− bone marrow cells was assessed 48 hours after retroviral transduction according to the manufacturer's instructions (APC-BrdU flow kit; BD PharMingen, San Diego, CA). Cells were incubated with 30 μM BrdU for 1 hour and stained with APC-conjugated anti-BrdU antibody and 7-AAD. Data were collected on a FACSCalibur (BD Bioscience) and analyzed by FlowJo software (TreeStar).
Results
Solution structure of the eTAFH domain-HEB peptide complex
Structural studies of apo-eTAFH were complicated by conformational exchange and/or aggregation at NMR concentrations, causing line-broadening and low signal-to-noise ratios for peaks in the NMR spectra. When eTAFH is complexed with the previously identified binding motif of the AD1 domain of HEB (HEB peptide; I11-S28),5 the poor signal-to-noise ratio and line-broadening were improved, making it possible to determine the eTAFH structure in a complex with the HEB peptide. However, no intermolecular NOEs were observed in half-filtered NOESY experiments, which prevented determination of the complex structure. Moderate affinity complexes such as this (Kd = 7 μM) undergo rapid chemical exchange, resulting in limited numbers of intermolecular NOEs. To address this, we fused the HEB peptide to the C-terminus of eTAFH with 14–amino acid linker between them. This resulted in a substantial increase in the local concentration of the peptide and, therefore, a decreased off-rate and longer residence time. We used the same approach to study the binding of the AML1-ETO MYND domain to a peptide derived from SMRT.13 The 15N-1H HSQC spectrum of the eTAFH-HEB fusion protein showed the resonances of eTAFH overlapped well with those of eTAFH in the noncovalent complex, but not with those in apo-eTAFH (Figure 1A). Subsequent dynamics measurements, including {1H}15N heteronuclear NOE, 15N R1, and 15N R2, showed that the linker is flexible, whereas the eTAFH and the HEB peptide portion of the fusion protein are relatively rigid with similar dynamic behavior (Figure 1B). 15N-1H HSQC comparison combined with the dynamics data indicate that the structure and binding mode are not disturbed in the covalently fused protein.

Binding mode is retained in the covalent eTAFH-HEB fusion protein. (A) Overlay of 15N-1H HSQC spectra of apo-eTAFH (red), eTAFH/HEB peptide complex (blue), and eTAFH-HEB fusion protein (green). The amides observed only when eTAFH is bound to HEB (![]() ), and the amides undergoing large chemical shift changes on HEB binding (→) are labeled with the residue number. *The peak for T321 is folded in the spectrum of eTAFH-HEB (green). (B) Backbone dynamics of eTAFH-HEB represented by plots of {1H}15N heteronuclear NOE and R2 relaxation rate versus residue number.
), and the amides undergoing large chemical shift changes on HEB binding (→) are labeled with the residue number. *The peak for T321 is folded in the spectrum of eTAFH-HEB (green). (B) Backbone dynamics of eTAFH-HEB represented by plots of {1H}15N heteronuclear NOE and R2 relaxation rate versus residue number.
Binding mode is retained in the covalent eTAFH-HEB fusion protein. (A) Overlay of 15N-1H HSQC spectra of apo-eTAFH (red), eTAFH/HEB peptide complex (blue), and eTAFH-HEB fusion protein (green). The amides observed only when eTAFH is bound to HEB (![]() ), and the amides undergoing large chemical shift changes on HEB binding (→) are labeled with the residue number. *The peak for T321 is folded in the spectrum of eTAFH-HEB (green). (B) Backbone dynamics of eTAFH-HEB represented by plots of {1H}15N heteronuclear NOE and R2 relaxation rate versus residue number.
), and the amides undergoing large chemical shift changes on HEB binding (→) are labeled with the residue number. *The peak for T321 is folded in the spectrum of eTAFH-HEB (green). (B) Backbone dynamics of eTAFH-HEB represented by plots of {1H}15N heteronuclear NOE and R2 relaxation rate versus residue number.
The solution structure of the eTAFH domain–HEB peptide complex was calculated using NOEs, chemical shifts, coupling constants, and residual dipolar couplings (RDCs) without any significant constraint violations (Table 1). An ensemble of 20 lowest-energy structures shows a well-defined eTAFH domain and HEB peptide (Figure 2A). The eTAFH domain is composed of 5 α-helices and a relatively long loop between helix 3 and helix 4. Helix 5 is locally but not globally well defined in the NMR ensemble because of a lack of long-range distance constraints, consistent with the increased motion of this helix seen in the dynamics measurements (Figure 1B). We generated a truncated mutant of eTAFH containing only the first 4 α-helices, and it showed the same affinity for the HEB peptide as the full-length eTAFH domain as assayed by ITC (data not shown). On the basis of the dynamics data and HEB-binding of truncated eTAFH, we designated residues A267 to Q353 as the functional eTAFH domain (Figure 2A-D). The linker between eTAFH and the HEB peptide is unstructured, as manifested by dynamics data (Figure 1B), sharp resonance lines, and the lack of NOEs. The HEB peptide adopts a short amphipathic α-helix spanning residues D14 to L21, a bend at D22 and F23, and one turn of helix for S25 to F28 (Figure 2B). Only residues from D14 to L21, including the first short helix, are well defined in the NMR ensemble and are interacting with eTAFH. Although residues from S25 to F28 are locally defined as one turn of helix, they are not superimposed in the ensemble because of the lack of intermolecular NOEs. {1H}15N heteronuclear NOE and R2 data show S25 to F28 is relatively flexible compared with D14 to D22, indicating that this part of HEB is not contacting eTAFH (Figure 1B). Our structure of the eTAFH domain generally agrees with previously reported structures (2H7B and 2PP4) and the TAFH domain of human TAF4 (2P6V), with the exception of the C-terminal half of helix 4.20,21,28
NMR restraints and statistics
| NMR constraints | Values |
|---|---|
| Distance constraints | 1851 |
| Intraresidual | 911 |
| Sequential | 483 |
| Medium-range | 196 |
| Long-range | 215 |
| Ambiguously assigned | 46 |
| Dihedral angle constraints* | |
| φ | 79 |
| ψ | 79 |
| JHNHα coupling constants | 20 |
| Residual dipolar couplings | |
| JHN | 66 |
| JNC′ | 50 |
| JC′Cα | 54 |
| RMS deviations for constraints | |
| Distance constraints, Å | 0.016 ± 0.002 |
| Dihedral angles, degrees | 0.33 ± 0.06 |
| JHNHα coupling constants (Hz) | 0.78 ± 0.05 |
| RDCs (Hz) | |
| JHN | 1.69 ± 0.09 |
| JNC′ | 0.34 ± 0.01 |
| JC′Cα | 0.95 ± 0.06 |
| RMS deviations from covalent geometry | |
| Bond lengths, Å | 0.003 ± 0.0002 |
| Bond angles, degrees | 0.45 ± 0.014 |
| Impropers, degree | 0.31 ± 0.017 |
| Ramachandran plot statistics (%)† | eTAFH (267-353), HEB (14-26) |
| Residues in most favored regions | 85.0 |
| Residues in additionally allowed regions | 12.0 |
| Residues in generously allowed regions | 2.1 |
| Residues in disallowed regions | 1.3 |
| RMS deviations from average structure, Å | eTAFH (270-345), HEB (16-21) |
| Backbone atoms | 0.77 ± 0.13 |
| Heavy atoms | 1.59 ± 0.16 |
| NMR constraints | Values |
|---|---|
| Distance constraints | 1851 |
| Intraresidual | 911 |
| Sequential | 483 |
| Medium-range | 196 |
| Long-range | 215 |
| Ambiguously assigned | 46 |
| Dihedral angle constraints* | |
| φ | 79 |
| ψ | 79 |
| JHNHα coupling constants | 20 |
| Residual dipolar couplings | |
| JHN | 66 |
| JNC′ | 50 |
| JC′Cα | 54 |
| RMS deviations for constraints | |
| Distance constraints, Å | 0.016 ± 0.002 |
| Dihedral angles, degrees | 0.33 ± 0.06 |
| JHNHα coupling constants (Hz) | 0.78 ± 0.05 |
| RDCs (Hz) | |
| JHN | 1.69 ± 0.09 |
| JNC′ | 0.34 ± 0.01 |
| JC′Cα | 0.95 ± 0.06 |
| RMS deviations from covalent geometry | |
| Bond lengths, Å | 0.003 ± 0.0002 |
| Bond angles, degrees | 0.45 ± 0.014 |
| Impropers, degree | 0.31 ± 0.017 |
| Ramachandran plot statistics (%)† | eTAFH (267-353), HEB (14-26) |
| Residues in most favored regions | 85.0 |
| Residues in additionally allowed regions | 12.0 |
| Residues in generously allowed regions | 2.1 |
| Residues in disallowed regions | 1.3 |
| RMS deviations from average structure, Å | eTAFH (270-345), HEB (16-21) |
| Backbone atoms | 0.77 ± 0.13 |
| Heavy atoms | 1.59 ± 0.16 |
Backbone dihedral angles from TALOS prediction.
Ramachandran plot statistics from PROCHECK-NMR analysis.

Solution structure of the eTAFH domain–HEB peptide complex. (A) Stereoview of an ensemble of 20 lowest energy NMR solution structures. The backbone of residues G267 to Q353 of the eTAFH domain of AML1-ETO (turquoise) and residues I12 to M26 of the AD1 domain from HEB (orange) are displayed after superimposing the structures using residues Q269 to L350 of eTAFH and E16-L21 of HEB. (B) Ribbon representation of the lowest energy structure. (C) Surface of the eTAFH domain with the side chains of the binding site displayed and labeled (white indicate nonpolar residues; red, acidic residues; blue, basic residues; green, polar residues). (D) HEB backbone represented as a ribbon with the side chains interacting with eTAFH domain displayed and labeled. Vmd-Xplor was used to generate the figures.
Solution structure of the eTAFH domain–HEB peptide complex. (A) Stereoview of an ensemble of 20 lowest energy NMR solution structures. The backbone of residues G267 to Q353 of the eTAFH domain of AML1-ETO (turquoise) and residues I12 to M26 of the AD1 domain from HEB (orange) are displayed after superimposing the structures using residues Q269 to L350 of eTAFH and E16-L21 of HEB. (B) Ribbon representation of the lowest energy structure. (C) Surface of the eTAFH domain with the side chains of the binding site displayed and labeled (white indicate nonpolar residues; red, acidic residues; blue, basic residues; green, polar residues). (D) HEB backbone represented as a ribbon with the side chains interacting with eTAFH domain displayed and labeled. Vmd-Xplor was used to generate the figures.
Basis for HEB binding to the eTAFH domain
Residues D14 to D22 of HEB, including 2 turns of α-helix, lie in the hydrophobic groove formed by helix 1, helix 4, and the loop between helix 3 and 4 of eTAFH (Figure 2B,D). The HEB peptide is anchored to the hydrophobic groove of eTAFH through its 3 conserved leucines: L17, L20, and L21 (Figure 2C,D). The short sequence containing the 3 leucines has the sequence LXXLL (L, leucine; X, any amino acid), a motif that is found in the E proteins E2A and E2-2 and in other protein–protein interfaces.5,29-32 L17 of HEB-LXXLL packs its side chain into a shallow groove formed by F277 and the T280 methyl of eTAFH. The bulky side chains of L20 and L21 are deeply embedded into 2 hydrophobic pockets established by F277, T280, L281, F284, L325, V329, F332, and L333. All 3 leucines of LXXLL wrap around the solvent-exposed side chain of F227 in eTAFH. The side chains of E16 of the HEB peptide and K273 of eTAFH are close enough for an electrostatic interaction, explaining the amino acid conservation at this position. The sequence and spacing of E16, L17, L20, and L21 determine the orientation of the HEB peptide in the eTAFH groove. The side chains of S18 and D19, which are the 2 X residues of the LXXLL motif, are solvent exposed and do not contact any eTAFH residues. The interaction between the HEB peptide and the binding groove of eTAFH buries a 714 Å2 solvent-accessible surface area of eTAFH, which is predominantly hydrophobic. This hydrophobic surface may explain the unfavorable behavior of apo-eTAFH in solution. The axis of the α-helix of HEB is tilted by approximately 72 degrees relative to the axis of helix 1 of eTAFH, which is the most notable difference from the docking models reported by the 2 other groups20,21 (Document S1, available on the Blood website; see the Supplemental Materials link at the top of the online article).
Mutations in the eTAFH domain that attenuate HEB binding
On the basis of the complex structure, we generated mutations in eTAFH that potentially disrupt the interaction between eTAFH and the HEB peptide to examine the importance of HEB association for the function of AML1-ETO. The binding affinities of the mutant eTAFH domains to the HEB peptide were measured by ITC and were compared with that of the wild-type eTAFH domain (Figure 3A). The side chain of F277 is located in the center of the hydrophobic pocket and participates in interactions with 3 leucines of the HEB peptide. A F277A mutation disrupted the fold of the eTAFH domain as assessed by comparison of the wild-type and F277A 15N-1H HSQC spectra (Figure 3B). 15N-1H HSQC spectra of the L325E and F332A mutants, however, showed little change compared with the wild-type protein, indicating that these mutations cause no significant change in the protein structure (data not shown). L325 is located in the loop between helix 3 and 4 and contacts L21 of HEB. The binding of the L325E mutant was impaired by 5-fold (Kd ∼34 μM), and a combined mutation of L325E/V329D did not further alter the affinity (data not shown). The eTAFH F332A mutant bound HEB very weakly, making it difficult to determine a Kd precisely. We could only approximate the Kd when the ITC data were fit to 1:1 binding with fixed stoichiometry, which showed that the affinity was decreased approximately 70-fold (Figure 3A). The affinity of the F332A mutant was also shown to be approximately 70-fold weaker by NMR titration experiments (data not shown).

eTAFH mutations impair HEB binding. (A) ITC measurements of the binding of HEB peptide to wild-type and mutant eTAFH domains. In each panel, the top portion is the raw data, and the bottom portion is a plot of the binding corrected for dilution enthalpy (squares indicate experimental data; line, fit to a one-site binding model). The average N (stoichiometry) and Kd from 2 independent experiments (± SD) for each protein are shown in the box. The stoichiometry was fixed to 1 to fit the F332A mutant data. (B) Overlay of 15N-1H HSQC spectra of wild-type eTAFH domain (red) and the F277A mutant (blue). Most peaks in the F277A mutant are absent or shifted, indicating the structure is disrupted.
eTAFH mutations impair HEB binding. (A) ITC measurements of the binding of HEB peptide to wild-type and mutant eTAFH domains. In each panel, the top portion is the raw data, and the bottom portion is a plot of the binding corrected for dilution enthalpy (squares indicate experimental data; line, fit to a one-site binding model). The average N (stoichiometry) and Kd from 2 independent experiments (± SD) for each protein are shown in the box. The stoichiometry was fixed to 1 to fit the F332A mutant data. (B) Overlay of 15N-1H HSQC spectra of wild-type eTAFH domain (red) and the F277A mutant (blue). Most peaks in the F277A mutant are absent or shifted, indicating the structure is disrupted.
eTAFH domain binds the (D/E)LXXLL motif of cMyb and STAT6
A careful examination of the binding interface in the complex structure showed that 4 residues (E16, L17, L20, and L21) of HEB are making the most significant contacts with the eTAFH domain (Figure 2D). To determine whether this putative motif is the binding determinant, we identified 3 different proteins (CBP, STAT6, and cMyb) that have a ζLXXLL (ζ, any polar residue) motif by a sequence search and assessed their binding to the eTAFH domain by NMR titration experiments. The peptides containing residues E292 to L309 of cMyb and L796 to S813 of STAT6 induced similar patterns of chemical shift changes as seen with HEB when titrated into a solution of the eTAFH domain, indicating that the cMyb and STAT6 peptides specifically bind eTAFH in a HEB-like manner (Figure 4A). The equilibrium dissociation constant (Kd) of the eTAFH/cMyb complex was calculated to be approximately 21 μM by fitting the titration curves (Figure 4B). The binding affinity of STAT6 for the eTAFH domain could not be accurately determined because we were unable to obtain saturation points in the titration curve, indicating that the binding is very weak. The N-CoR R1 domain was previously identified as a binding partner for the eTAFH domain,21,33 so we quantified its binding affinity as well (Figure 4B). Sequence alignment indicated that only the 3 leucines and the −1 residue of the LXXLL motif are conserved in the proteins that bind eTAFH (Figure 4D). This result agrees well with our complex structure, in which those 4 residues appear to mediate the critical contacts for binding eTAFH. An acidic residue seems to be required at the −1 position of the LXXLL motif for eTAFH binding because the CBP LXD domain, which has a glutamine at this position, did not bind to eTAFH. Given that the affinities of eTAFH for HEB and cMyb are higher than for STAT6, a glutamic acid is apparently preferred at this position. This preference can be attributed to the interaction between the longer side chain of glutamic acid with K273 of eTAFH that we observed in the complex structure. N-CoR has 3 isoleucines instead of leucines in an ELXXLL motif, and this may explain its relatively weak affinity for eTAFH (Kd ∼75 μM) compared with HEB and cMyb. Taken together, (D/E)φXXφφ (φ, hydrophobic) is the minimum requirement for eTAFH binding with leucines preferred at the φ position.

eTAFH domain binds peptides with a conserved (D/E)LXXLL motif from HEB, cMyb, N-CoR, and STAT6. (A) Overlays of selected amides in the 15N-1H HSQC spectra of the eTAFH domain recorded as a function of the concentration of each peptide. Each column represents titration data for 3 separate amides with one particular peptide. (B) Kd determination using chemical shift changes resulting from titrations of each peptide. The 3 best fits from changes in 1HN shifts for each peptide are displayed. (C) Results of a Kd determination using fluorescence polarization with 0.2 μM fluorescein-labeled HEB peptide (FLSN-TDKELSDLLD) and increasing concentrations of eTAFH. Results of one titration are shown. Two independent experiments were carried out resulting in Kd = 12.5 ± 2.1 μM. (D) Sequence alignment of peptides examined for eTAFH binding. Consensus amino acids are colored in blue. Asterisks indicate residues in HEB whose side chains contact the eTAFH domain.
eTAFH domain binds peptides with a conserved (D/E)LXXLL motif from HEB, cMyb, N-CoR, and STAT6. (A) Overlays of selected amides in the 15N-1H HSQC spectra of the eTAFH domain recorded as a function of the concentration of each peptide. Each column represents titration data for 3 separate amides with one particular peptide. (B) Kd determination using chemical shift changes resulting from titrations of each peptide. The 3 best fits from changes in 1HN shifts for each peptide are displayed. (C) Results of a Kd determination using fluorescence polarization with 0.2 μM fluorescein-labeled HEB peptide (FLSN-TDKELSDLLD) and increasing concentrations of eTAFH. Results of one titration are shown. Two independent experiments were carried out resulting in Kd = 12.5 ± 2.1 μM. (D) Sequence alignment of peptides examined for eTAFH binding. Consensus amino acids are colored in blue. Asterisks indicate residues in HEB whose side chains contact the eTAFH domain.
There are some notable discrepancies in the binding motif between the modeling studies and the structure. Transactivation activity and eTAFH engagement of HEB were lost in reporter gene and yeast 2-hybrid assays when F23 of HEB was substituted with alanine or arginine.5 However, there is little similarity at the F23 position among eTAFH-binding peptides (Figure 4D), and a truncated HEB peptide excluding F23 was sufficient for eTAFH engagement (Figure 4C). It was assumed that the acidic residues D19 and D22 of HEB would provide additional surfaces specific for eTAFH as opposed to CBP/p300, based on reporter gene and 2-hybrid assays.5 However, the side chains of D19 and D22 do not contact eTAFH in the complex structure, and neither aspartic acid residue is conserved in cMyb and STAT6 (Figure 4D). It is possible that the 2 aspartic acid residues help to establish amphipathicity of the peptide rather than make direct contacts with eTAFH.
Contribution of the eTAFH domain–HEB interaction to AML1-ETO's ability to inhibit granulocyte differentiation
Previous studies showed that AML1-ETO can repress granulocyte differentiation in an established hematopoietic cell line or in primary mouse bone marrow cells.10,13 To test the contributions of the eTAFH domain–E protein interaction for the repressive activity of AML1-ETO, we transduced primary, lineage-depleted (CD5−, B220−, Mac-1−, Gr-1−, Ter119−, Lin−) mouse bone marrow cells with retroviruses expressing green fluorescent protein (GFP) alone (MigR1), AML1-ETO, or AML1-ETO proteins with single amino acid substitutions in the eTAFH domain (Figure 5A) and assessed granulocyte differentiation after 7 days of culture in the presence of IL-3, IL-6, SCF, and G-CSF in the successfully transduced (GFP+) cells. Neither the F332A mutation that decreases the binding of eTAFH to the HEB peptide by 70-fold nor the F277A mutation that disrupts the eTAFH domain structure significantly affected the activity of AML1-ETO to repress granulocyte differentiation as measured by the percentage of Gr-1+ Mac-1+ cells after 7 days of in vitro culture (Figure 5B). We were previously able to show a weak activity conferred by the MYND domain by combining a MYND mutation with a 7–amino acid substitution in the HHR domain (m7) that disrupts oligomerization6,13 ; therefore, we also assessed the effects of the eTAFH domain mutations in this context. As we showed previously,6 the m7 mutation partially ameliorated AML1-ETO's inhibition of granulocyte differentiation (Figure 5C,D). However, neither the F277A nor F332A mutations, when combined with the m7 mutation, significantly improved granulocyte differentiation compared with the m7 mutation alone.

In vivo activities of AML1-ETO are relatively unaffected by mutations that impair HEB binding. (A) Schematic diagram of AML1-ETO and the location of mutations (asterisks). (B) Effect of eTAFH mutations on AML1-ETO's repression of granulocyte differentiation after 7 days of culture in the presence of IL-3, IL-6, SCF, and G-CSF. Cells within the forward and side scatter gates were further gated for GFP expression, and GFP-positive cells were examined for Mac-1 and Gr-1 expression. Data represent triplicate samples from 2 independent experiments. Error bars represent 95% confidence intervals. Significant differences from AML1-ETO (#) are indicated with asterisks (ANOVA and Dunnett multiple comparison test, **P < .01). (C) Representative flow of Lin− bone marrow cells infected with MigR1 retroviruses expressing AML1-ETO, the m7 oligomerization mutant, and the m7+eTAFH mutants. (D) Average percentages of Gr-1+Mac-1+ cells. Significant differences relative to the m7 mutant are indicated with asterisks (ANOVA and Dunnett multiple comparison test, **P < .01, *P < .05). (E) Cos7 cells were cotransfected with AML1/ETO and its mutated derivatives and FLAG-tagged HEB. (Top) Cell lysates immunoprecipitated (IP) with anti-FLAG and blotted with antibody to the Runt domain (RD) in AML1/ETO. (Middle) Input lysate (1%) was blotted with anti-RD to detect AML1/ETO proteins. (Bottom) Membranes from the top panel were reprobed with anti-FLAG antibodies. The percentages of immunoprecipitated AML1-ETO proteins relative to input were 3.7% (AML1/ETO), 2.3% (AML1-ETO F332A), 1.1% (AML1-ETO m7), and 0.7% (AML1-ETO m7+F332A). (F) Representative flow of BrdU incorporation 48 hours after transduction of Lin− bone marrow cells with MigR1 expressing GFP, AML1-ETO, or the AML1-ETO eTAFH mutants. (G) Percentage of GFP+ cells that had incorporated BrdU after an 1-hour BrdU pulse. Data are from 2 experiments each with triplicate samples (error bars = 95% confidence intervals; significant differences from AML1-ETO indicated with an asterisk, ANOVA and Dunnett multiple comparison test, **P < .01). (H) Serial replating of bone marrow cells. Graphs represent the average number of colonies from each round of replating in the presence of IL-3, IL-6, and SCF. Day 7 represents colony numbers per 103 cells plated and days 14, 21, and 28 from 104 plated cells. Numbers are averaged from 3 experiments, each containing triplicate samples. The numbers of colonies derived from F277A- and F332A-transduced cells were significantly lower than those from AML1-ETO–transduced cells at day 14 and day 28 (P < .01). Error bars represent 95% confidence intervals.
In vivo activities of AML1-ETO are relatively unaffected by mutations that impair HEB binding. (A) Schematic diagram of AML1-ETO and the location of mutations (asterisks). (B) Effect of eTAFH mutations on AML1-ETO's repression of granulocyte differentiation after 7 days of culture in the presence of IL-3, IL-6, SCF, and G-CSF. Cells within the forward and side scatter gates were further gated for GFP expression, and GFP-positive cells were examined for Mac-1 and Gr-1 expression. Data represent triplicate samples from 2 independent experiments. Error bars represent 95% confidence intervals. Significant differences from AML1-ETO (#) are indicated with asterisks (ANOVA and Dunnett multiple comparison test, **P < .01). (C) Representative flow of Lin− bone marrow cells infected with MigR1 retroviruses expressing AML1-ETO, the m7 oligomerization mutant, and the m7+eTAFH mutants. (D) Average percentages of Gr-1+Mac-1+ cells. Significant differences relative to the m7 mutant are indicated with asterisks (ANOVA and Dunnett multiple comparison test, **P < .01, *P < .05). (E) Cos7 cells were cotransfected with AML1/ETO and its mutated derivatives and FLAG-tagged HEB. (Top) Cell lysates immunoprecipitated (IP) with anti-FLAG and blotted with antibody to the Runt domain (RD) in AML1/ETO. (Middle) Input lysate (1%) was blotted with anti-RD to detect AML1/ETO proteins. (Bottom) Membranes from the top panel were reprobed with anti-FLAG antibodies. The percentages of immunoprecipitated AML1-ETO proteins relative to input were 3.7% (AML1/ETO), 2.3% (AML1-ETO F332A), 1.1% (AML1-ETO m7), and 0.7% (AML1-ETO m7+F332A). (F) Representative flow of BrdU incorporation 48 hours after transduction of Lin− bone marrow cells with MigR1 expressing GFP, AML1-ETO, or the AML1-ETO eTAFH mutants. (G) Percentage of GFP+ cells that had incorporated BrdU after an 1-hour BrdU pulse. Data are from 2 experiments each with triplicate samples (error bars = 95% confidence intervals; significant differences from AML1-ETO indicated with an asterisk, ANOVA and Dunnett multiple comparison test, **P < .01). (H) Serial replating of bone marrow cells. Graphs represent the average number of colonies from each round of replating in the presence of IL-3, IL-6, and SCF. Day 7 represents colony numbers per 103 cells plated and days 14, 21, and 28 from 104 plated cells. Numbers are averaged from 3 experiments, each containing triplicate samples. The numbers of colonies derived from F277A- and F332A-transduced cells were significantly lower than those from AML1-ETO–transduced cells at day 14 and day 28 (P < .01). Error bars represent 95% confidence intervals.
The F332A mutation decreased the interaction of AML1-ETO and the AML1-ETO m7 mutant protein with HEB by approximately 2-fold and approximately 5-fold, respectively, as assessed by coimmunoprecipitation of ectopically expressed full-length proteins in Cos7 cells (Figure 5E). These modest perturbations in binding could be due to additional HEB interaction sites on the full-length AML1-ETO protein, or through the binding of HEB to other proteins that directly interact with full-length AML1-ETO.
Contribution of the eTAFH domain–HEB interaction to AML1-ETO's ability to inhibit short-term proliferation and promote long-term self-renewal
AML1-ETO inhibits the short-term proliferation of human and mouse primary bone marrow cells.34,35 We introduced full-length AML1-ETO and its mutated derivatives into Lin− primary mouse bone marrow cells and measured proliferation 2 days later by BrdU incorporation. Neither the F277A nor the F332A mutations released the proliferation block imposed by AML1-ETO (Figure 5F,G).
Another well-characterized activity of AML1-ETO is that it increases the clonogenicity of primary bone marrow cells, and as a result AML1-ETO–transduced cells can be serially replated in vitro.34-36 This self-renewal activity is considered to be pertinent to the oncogenic activity of AML1-ETO. We transduced Lin− BM cells with retroviruses expressing AML1-ETO or its mutated derivatives and cultured them in methylcellulose medium supplied with IL-3, IL-6, and SCF. Neither the F277A nor the F332A mutations eliminated AML1-ETO's ability to confer increased self-renewal capacity on hematopoietic progenitors in vitro (Figure 5H), although the number of colonies regenerated at each round of replating was significantly lower, suggestive of a minor decrease in protein stability or activity or both.
Discussion
Previous studies identified E2A, E2-2, and HEB as proteins that bind to the eTAFH domain in AML1-ETO. It was proposed that the dysregulation of E protein target genes by recruitment of AML1-ETO may be important for t(8;21) leukemogenesis.5,37 Here, we solved the structure of the eTAFH domain of AML1-ETO with an interacting peptide from HEB. With the use of this structural information, we show that the interaction between the eTAFH domain of AML1-ETO and the E proteins contributes relatively little to the activity of AML1-ETO. Our results are consistent with a recent study in which deletion of the eTAFH domain from AML1-ETO9a, which induces leukemia by itself, was shown to have no effect on the ability of the protein to induce leukemia in mice.38 We cannot, however, rule out a contribution of E proteins to AML1-ETO–mediated leukemogenesis because a mutation that severely impairs binding of the isolated eTAFH domain to HEB did not eliminate all HEB binding to the full-length AML1-ETO protein.
We have shown that the LXXLL motif of HEB (with the preceding residue Glu) is recognized by eTAF domain of AML1-ETO. LXXLL motifs are also found in nuclear hormone receptor (NR)–interacting cofactors, such as p160/SRC/NCoA,29-31 and in transcription factors (TFs), such as STAT6 and cMyb. Most LXXLL motifs in NR–cofactor interactions have a hydrophobic or basic residue at the −1 position. However, the −1 residues of LXXLL motifs found in TFs are acidic as in HEB, and these motifs mediate interaction with the coactivators CBP/P300 or NCoA-1.5,32,39,40 The LXXLL motif in HEB shows similarities to the LXXLL motifs of cMyb and STAT6. NCoA-1 binding of STAT6 is mediated by a 9-residue long α-helix formed by E799 to L807, analogous to the interaction of HEB residues T13 to L21 with eTAFH. Only the side chains of DLXXLL on the STAT6 helix are facing the binding interface, similar to ELXXLL of HEB.41 A relatively long cMyb fragment mediates the interaction with CBP/P300, but a bend similar to that in HEB is observed in the α-helix of cMyb at S304, enabling the side chain of L302 to penetrate deeply into the binding pocket. Taking all 3 complex structures together, we found that the LXXLL motifs of TFs and HEB adopt a characteristic orientation to interact with target proteins, which is comparable to LXXLL motifs of coactivators that bind NRs. The orientation of the LXXLL motif in NR–coactivator interaction is determined by a charge clamp assisted by hydrogen-bonds between lysine and glutamic acid of the ligand binding domain (LBD) of NR and the backbone of the LXXLL motif, as well as hydrophobic interactions (Figure 6A).31,42-44 On the contrary, no such “charge clamp” was found in the interactions mediated by the LXXLL motifs of transcription factors. Instead, the N-terminal flanking charged residue of the LXXLL motif interacts electrostatically with a charged side chain of target proteins, determining the orientation in concert with hydrophobic interactions (Figure 6B-D). Therefore, those specific interactions mediated by each class of LXXLL motifs generate almost opposite orientations between the 2 types of interaction (Figure 6A-D).

Cartoon representations of the orientation of LXXLL motifs found in 2 different classes. (A) The orientation of LXXLL motifs of coactivators on the ligand-binding domain of nuclear receptors (NRs). (B-D) The binding orientation of LXXLL motifs of transcription factors: (B) STAT6 binding the PAS-B domain of NcoA-1, (C) cMyb binding the KIX domain of CBP, (D) HEB binding the eTAFH domain of AML1-ETO. The glutamic acids of STAT6 and HEB and the arginine of cMyb providing side chain interactions are in light gray and labeled.
Cartoon representations of the orientation of LXXLL motifs found in 2 different classes. (A) The orientation of LXXLL motifs of coactivators on the ligand-binding domain of nuclear receptors (NRs). (B-D) The binding orientation of LXXLL motifs of transcription factors: (B) STAT6 binding the PAS-B domain of NcoA-1, (C) cMyb binding the KIX domain of CBP, (D) HEB binding the eTAFH domain of AML1-ETO. The glutamic acids of STAT6 and HEB and the arginine of cMyb providing side chain interactions are in light gray and labeled.
The repression of E protein activity was proposed as an alternative model for AML1-ETO's transcriptional repression mechanism that is independent of its RUNX1 DNA binding activity.45 E proteins positively regulate genes that are essential for hematopoiesis by interacting with E-box elements within their promoters, whereby they recruit the transcriptional coactivators p300/CBP histone acetyltransferases.5 It was proposed that in t(8;21) cells, expression of these E protein target genes may be silenced by a dominant interaction of E proteins with AML1-ETO that precludes promoter occupancy by p300/CBP and facilitates occupancy by HDAC-containing complexes.
The ability of leukemia-associated transcription factors to confer serial replating capacity to primary bone marrow cells correlates well with their transforming properties.46-49 Disrupting the HEB binding site in the eTAFH domain did not eliminate the ability of AML1-ETO to confer self-renewal capacity for up to 4 weeks. However, disrupting AML1-ETO oligomerization through mutations in HHR,6 or by impairing either DNA or CBFβ binding by the Runt domain50 completely eliminated the ability of AML1-ETO to confer serial replating to primary bone marrow cells. It is possible that oligomerization through the HHR domain might mask the effects of mutations in eTAFH by mediating the formation of mixed tetramers with endogenous wild-type ETO or its homologues.6 However, specifically disrupting the eTAFH–E protein interaction in the context of mutations in HHR that impaired oligomerization did not significantly affect the deleterious effects of AML1-ETO on the granulocyte differentiation of primary mouse bone marrow cells, suggesting that recruitment of E proteins by other ETO homologues cannot explain the relatively small effect of the eTAFH mutations. Thus far, the interactions of AML1-ETO with DNA, with CBFβ, and with itself or other ETO proteins through oligomerization appear to be the most important properties of AML1-ETO, and therefore the promising potential targets for AML1-ETO inhibitors.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported by the National Institutes of Health (NIH; grant R01 CA108056) (J.H.B.). NMR data at 900 MHz were collected at the National Magnetic Resonance Facility at Madison, which is supported by NIH (grants P41RR02301 [BRTP/NCR]) and P41GM66326 [NIGMS]).
National Institutes of Health
Authorship
Contribution: S.P., W.C., X.C., and T.C. designed and executed the experiments; M.T. collected NMR data at 900 MHz; and N.A.S. and J.H.B. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: John H. Bushweller, Dept of Molecular Physiology and Biological Physics, PO Box 800736, UVa Health Sciences Center, Charlottesville, VA 22908; e-mail: jhb4v@virginia.edu; and Nancy A. Speck, Dept of Cell and Developmental Biology and Abramson Family Cancer Research Institute, 510 BRB II/III, University of Pennsylvania, Philadelphia, PA 19104; e-mail: nancyas@exchange.upenn.edu.
