

Abstract
Activation of the Wnt/β-catenin signaling pathway is a hallmark of a number of solid tumors. We analyzed the regulation of the Wnt/β-catenin pathway in acute lymphoblastic leukemia (ALL) and its role in the pathogenesis of the disease. We found that expression of the Wnt inhibitors sFRP1, sFRP2, sFRP4, sFRP5, WIF1, Dkk3, and Hdpr1 was down-regulated due to abnormal promoter methylation in ALL cell lines and samples from patients with ALL. Methylation of Wnt inhibitors was associated with activation of the Wnt-signaling pathway as demonstrated by the up-regulation of the Wnt target genes WNT16, FZ3, TCF1, LEF1, and cyclin D1 in cell lines and samples and the nuclear localization of β-catenin in cell lines. Treatment of ALL cells with the Wnt inhibitor quercetin or with the demethylating agent 5-aza-2′-deoxycytidine induced an inactivation of the Wnt pathway and induced apoptosis of ALL cells. Finally, in a group of 261 patients with newly diagnosed ALL, abnormal methylation of Wnt inhibitors was associated with decreased 10-year disease-free survival (25% versus 66% respectively, P < .001) and overall survival (28% versus 61% respectively, P = .001). Our results indicate a role of abnormal Wnt signaling in ALL and establish a group of patients with a significantly worse prognosis (methylated group).
Introduction
The evolutionarily conserved Wnt-signaling pathway has pivotal roles during the development of many organ systems. Central to this pathway is a multiprotein scaffold consisting of adenomatous polyposis coli (APC), glycogen synthase kinase (GSK)–3β, axin, and the transcriptional cofactor β-catenin. In the absence of Wnt ligands, β-catenin levels are kept low through constitutive phosphorylation by GSK-3β, which leads to the ubiquitination and degradation of β-catenin.1-3 Binding of the Wnt ligands to their receptors, Frizzled (Fz), leads to activation of the adaptor protein, Disheveled (Dvl), and the inhibition of GSK-3β activity, reducing phosphorylation and subsequent degradation of β-catenin. β-Catenin is stabilized and translocates to the nucleus where it binds members of the T-cell factor (Tcf)/lymphoid-enhancing factor (Lef) family of transcription factors and induces the expression of target gene such as CMYC and Cyclin D1.4 This signaling pathway is controlled by a number of natural Wnt antagonists that interfere with ligand-receptor interactions or Dvl protein, including members of the Dickkopf (DKK) family, the secreted Frizzled-related protein (sFRP) family, the Wnt inhibitory factor-1 (WIF1), and the human Dapper protein-1 (HDPR1)
Recent studies have demonstrated a role for Wnt signal transduction at several stages of lymphocyte development and in the self-renewal of hematopoietic stem cells.5,6 All of them indicate a crucial role for the Wnt-signaling pathway in the proliferation of thymocytes and also of pro-B cells, suggesting that Wnt proteins can function as growth factors for progenitor cells of both the B- and T-cell lineages.4,7,8
Abnormal Wnt signaling has become a hallmark of some types of solid tumors, most notably colon and hepatocellular carcinomas,9,10 and has been commonly associated with mutations in the amino-terminal region of β-catenin that make the molecule resistant to processing through the degradation pathway or in other proteins in this pathway, like the APC gene.11 In addition, the functional loss of Wnt antagonists can contribute to activation of the Wnt pathway and result in carcinogenesis through deregulation of cell proliferation and differentiation. Recent studies have shown that impaired regulation of Wnt-antagonists such as sFRP, WIF1, HDPR1, and DKK3 by promoter hypermethylation is present in several human malignancies.12-17
Given that Wnt signals are important for the survival and expansion of lymphocyte progenitors, it has been suggested that dysregulated Wnt signaling could be a mechanism underlying lymphoid leukemogenesis. Recent studies in acute or chronic lymphoid leukemias have provided initial information implicating a role for Wnt signaling in malignant hematopoiesis. B cells of patients with chronic lymphocytic leukemia (CLL) have been shown to express several Wnt genes highly in proportion to normal B cells.18 In addition, in pre-B acute lymphoblastic leukemia (ALL), several abnormal fusion proteins can be present. The presence of the E2A-PBX1 fusion protein has been shown to induce the expression of Wnt-16.19
We have recently shown that the methylation of cytosine nucleotides in ALL cells may be the most important way of inactivating cancer-related genes in this disease. In fact, this epigenetic event can help to inactivate tumor-suppressive apoptotic or growth-arresting responses and has prognostic impact in B- and T-ALL.20-22 The presence in individual tumors of multiple simultaneously methylated genes is an independent factor in poor prognosis in both childhood and adult ALL in terms of both disease-free survival (DFS) and overall survival (OS). Interestingly, the DKK3 gene, which negatively modulates WNT7A signaling, is frequently silenced by methylation in ALL.15,23 It can thus be speculated that the functional loss of Wnt antagonists can contribute to activation of the Wnt pathway in ALL and may play a role in the pathogenesis and prognosis of the disease; however, this hypothesis has never been previously explored.
In this study, we demonstrate that silencing of the Wnt antagonists by promoter methylation contributes to constitutive activation of the Wnt-signaling pathway in ALL and that activation of the Wnt-signaling pathway is implicated in the pathogenesis of the disease.
Materials and methods
Cell lines and patients
Six ALL-derived cell lines (TOM-1, NALM-20, MY, LOUCY, JURKAT, and TANOUE) were purchased from the Deutche Sammlung von Microorganismen und Zellkulturen [DSMZ], Braunschweig, Germany). Cells were grown at 37°C under 5% CO2 in humidified air in RPMI medium (Gibco-BRL, Burlington, ON, Canada) supplemented with 20% fetal bovine serum (Gibco, Grand Island, NY), 1% penicillin/streptomycin and 1% HEPES (Gibco-BRL).
We studied 261 consecutive patients (154 male; 107 female) with de novo ALL who were enrolled in successive multicenter studies of the “Programa para el estudio y tratamiento de las hemopatias malignas” (PETHEMA) Spanish study group. All these patients were referred to the Reina Sofia Hospital of Cordoba, Spain, from January 1990 to December 2004. The median age at diagnosis in the study population as a whole was 14 years (range, 0.5-82 years). Of these patients, 129 were children (median age, 5 years; range, 0.5-14 years) and 132 presented adult ALL (median age, 29 years; range, 15-82 years). Informed consent was obtained from the patient or the patient's guardians, in accordance with the Declaration of Helsinki. This study was approved by the ethics committee at the University of Navarra. Diagnosis was established according to standard morphologic, cytochemical, and immunophenotypic criteria. Patients were studied at the time of initial diagnosis, were risk-stratified according to the therapeutic protocol used, which was always based on recognized prognostic features (including cytogenetics), and were entered in ALL protocols of the PETHEMA group. For statistical analyses, children were also grouped according to the National Cancer Institute (NCI) risk-classification criteria.24 The specific PETHEMA ALL treatment protocols in which these patients entered included ALL-89 (between 1990-1993; n = 51) and ALL-93 (between 1993-2004; n = 210). The design and results of these studies have been previously reported.25-27 PETHEMA ALL-89 consisted of a 5-drug induction therapy, followed by 4 cycles of early postremission treatment during 4 months, and maintenance therapy for 2 years. Patients in remission at the end of the first year were randomized to receive one 6-week cycle of late intensification therapy. PETHEMA ALL-93 consisted of a standard 5-drug/5-week induction course. Patients in complete remission (CR) with an HLA-identical family donor were assigned to allogeneic stem cell transplantation and the remaining patients were randomized to autologous transplantation or to delayed intensification followed by maintenance chemotherapy for up to 2 years after complete remission. Relapses occurred in 106 patients. Forty-five patients underwent stem cell transplantation (SCT; 15 autologous, 30 allogeneic) in the first (n = 20) or second CR (n = 25). There are 124 patients currently alive. The median follow-up of the series was 36 months. Clinical characteristics of the patients are listed in Table 1.
Clinical characteristics and outcome of 261 ALL patients according to gene methylation status
| Feature | Nonmethylated,no. patients | Methylated,no. patients | P |
|---|---|---|---|
| Total | 100 | 161 | |
| Age | NS | ||
| Younger than 15 y | 43 | 57 | |
| Older than 15 y | 35 | 65 | |
| Sex (M/F) | 60/40 | 61/39 | NS |
| WBC count | NS | ||
| Less than 50 × 109/L | 75 | 70 | |
| More than 50 × 109/L | 25 | 30 | |
| FAB classification | NS | ||
| L1 | 39 | 32 | |
| L2 | 49 | 57 | |
| L3 | 12 | 11 | |
| Blast lineage | NS | ||
| B cell | 91 | 89 | |
| T cell | 9 | 11 | |
| NCI risk groups | NS | ||
| Standard | 79 | 66 | |
| Poor | 21 | 34 | |
| PETHEMA risk groups | NS | ||
| Standard | 41 | 34 | |
| Poor | 59 | 66 | |
| Treatment | NS | ||
| PETHEMA 89 | 20 | 19 | |
| PETHEMA 93 | 80 | 81 | |
| BMT | 19 | 16 | NS |
| Best response | |||
| CR | 87 | 93 | NS |
| Cytogenetic/molecular abnormalities | NS | ||
| BCR-ABL | 17 | 14 | |
| t(1;19) | 4 | 2 | |
| 11q23 | 3 | 3 | |
| c-myc | 6 | 8 | |
| 7q35-14q11 | 6 | 8 | |
| Hyperdiploidy | 9 | 5 | |
| TEL-AML1 | 5 | 3 | |
| Normal | 40 | 48 | |
| Others | 3 | 3 | |
| NT | 7 | 6 | |
| Relapse | 29 | 59 | < .001 |
| Death | 36 | 59 | .001 |
| Feature | Nonmethylated,no. patients | Methylated,no. patients | P |
|---|---|---|---|
| Total | 100 | 161 | |
| Age | NS | ||
| Younger than 15 y | 43 | 57 | |
| Older than 15 y | 35 | 65 | |
| Sex (M/F) | 60/40 | 61/39 | NS |
| WBC count | NS | ||
| Less than 50 × 109/L | 75 | 70 | |
| More than 50 × 109/L | 25 | 30 | |
| FAB classification | NS | ||
| L1 | 39 | 32 | |
| L2 | 49 | 57 | |
| L3 | 12 | 11 | |
| Blast lineage | NS | ||
| B cell | 91 | 89 | |
| T cell | 9 | 11 | |
| NCI risk groups | NS | ||
| Standard | 79 | 66 | |
| Poor | 21 | 34 | |
| PETHEMA risk groups | NS | ||
| Standard | 41 | 34 | |
| Poor | 59 | 66 | |
| Treatment | NS | ||
| PETHEMA 89 | 20 | 19 | |
| PETHEMA 93 | 80 | 81 | |
| BMT | 19 | 16 | NS |
| Best response | |||
| CR | 87 | 93 | NS |
| Cytogenetic/molecular abnormalities | NS | ||
| BCR-ABL | 17 | 14 | |
| t(1;19) | 4 | 2 | |
| 11q23 | 3 | 3 | |
| c-myc | 6 | 8 | |
| 7q35-14q11 | 6 | 8 | |
| Hyperdiploidy | 9 | 5 | |
| TEL-AML1 | 5 | 3 | |
| Normal | 40 | 48 | |
| Others | 3 | 3 | |
| NT | 7 | 6 | |
| Relapse | 29 | 59 | < .001 |
| Death | 36 | 59 | .001 |
Data are expressed as percentages of patient population, except where otherwise noted.
NS indicates not significant; WBC, white blood cell; FAB, French-American-British; BMT, bone marrow transplantation; NT, not tested.
Methylation-specific PCR
Bone marrow samples were obtained from all the patients at the time of diagnosis. High-molecular-weight DNA was prepared from mononuclear diagnostic marrow cells using conventional methods, frozen at −80°C, and retrospectively analyzed to assess the role of methylation profile. In every case, the diagnostic bone marrow sample contained blast cells in a ratio of at least 70%. Methylation status of the CpG islands in the sFRP1, sFRP2, sFRP4, sFRP5, WIF1, DKK3, and HDPR1 gene promoters was determined by gDNA bisulfite treatment followed by methylation-specific PCR (MSP) as reported by Herman et al.28 Primer sequences of each gene for the unmethylated and methylated reactions have been reported elsewhere.12-16 “Hot start” PCR was performed for 30 cycles consisting of denaturation at 95°C for 1 minute, annealing at 60°C for 1 minute, and extension at 72°C for 1 minute, followed by a final 7-minute extension for all primer sets. The products were separated by electrophoresis on 2% agarose gel. Bone marrow mononuclear cell and peripheral lymphocyte DNAs from healthy donors were used as negative control for methylation-specific assays. Human male gDNA universally methylated for all genes (Intergen, Purchase, NY) was used as a positive control for methylated alleles. Water blanks were included with each assay. The sensitivity of the MSP was established by using totally methylated positive control DNA serially diluted by normal lymphocyte DNA. MSPs with 1:10, 1:100, and 1:1000 diluted positive control DNA produced detectable methylated bands (data not shown). Results were always confirmed by repeat MSP assays after an independently performed bisulfite treatment. Freezing and thawing of the DNA samples did not affect the MSP results (data not shown).
Expression analyses and quantitative real-time PCR
Expression of Wnt-antagonists (SFRP1, SFRP2, SFRP4, SFRP5, DKK3, WIF1, and HDPR1) and Wnt pathway genes (WNT16, FZ3, LEF1, TCF1, and cyclin D1) were analyzed by the quantitative real-time PCR technique (qRT-PCR). Total RNA was extracted from marrow samples with Ultraspec (Biotecx, Houston, TX) following the manufacturer's instructions. Reverse transcription was performed on 1 μg total RNA, after heating at 70°C for 5 minutes, with random hexamers as reaction primer. The reaction was carried out at 42°C for 45 minutes in the presence of 12 U avian myeloblastosis virus reverse transcriptase (Boehringer-Mannheim, Mannheim, Germany). qRT-PCR for gene expression was performed with the LightCycler technology, using 1 μL cDNA in 20 μL reaction volume with 0.4 μM each primer and 2 μL of 10 × LightCycler FastStar DNA Master SYBR Green I (Roche Molecular Biochemicals, Mannheim, Germany). The final Mg2+ concentration in the reaction mixture was adjusted to 3.5 mM. Primers for each gene have been previously reported.18 The following program conditions were applied for qRT-PCR running: denaturation program, consisting of one cycle at 95°C for 8 minutes; amplification program, consisting of 45 cycles at 95°C for 5 seconds, 60°C for 10 seconds, and 72°C for 15 seconds; melting program, one cycle at 95°C for 0 seconds, 40°C for 60 seconds, and 90°C for 0 second; and cooling program, one cycle at 40°C for 60 seconds. The temperature transition rate was 20°C/s, except in the melting program, which was 0.4°C/s between 40°C and 90°C. Abelson gene (ABL1) was used as reference gene, and it was amplified in the same run and following the same procedure described (forward: 5′-CCCAACCTTTTCGTTGCACTGT-3′; reverse: 5′-CGGCTCTCGGAGGAGACGTAGA-3′). To reduce the variation between different assays and samples, a procedure based on the relative quantification of target genes versus their controls/calibrators in relation to the reference gene was used. Calculations were automatically performed by LightCycler software (RealQuant, version 1.0, Roche). The normalized ratios (N) for each gene were obtained from the next equation and expressed as percentage of the control/calibrator:
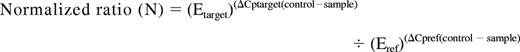
Efficiencies (E) of each gene were calculated from the slopes of crossover points (Cp) versus DNA concentration plot, according to the formula: E = 10(−1/slope). ΔCp corresponded to the difference between control/calibrator Cp and sample Cp, either for the target or for the reference sequences. The selected controls/calibrators were normal lymphocytes from healthy donors. They were considered as 100% expression.
Treatment with the demethylating agent 5-aza-2′-deoxycytidine
ALL-derived TOM-1 and NALM-20 cell lines were grown at a density of 600 000 cells/mL in 25-cm2 flasks with 10 mL RPMI 1640 medium supplemented with 20% fetal bovine serum and maintained at 37°C in a humid atmosphere containing 5% CO2. Cell lines were treated with 2 and 4 μM 5-aza-2′-deoxycytidine (Sigma-Aldrich, Steinheim, Germany) for 4 days. After treatment, cells were washed in PBS, pelleted by centrifugation at 1000 g during 5 minutes and used for gDNA and RNA isolation. DNA was extracted using QIAmp DNA Mini Kit (Qiagen, Hilden, Germany) and total RNA using Rneasy Mini Kit (Qiagen). Total RNA (1 μg) was used for cDNA synthesis using SuperScript II RNase H-RT (Invitrogen Life Technologies, Paisley, United Kingdom) with random hexamers.
Protein extraction
Cell pellets were resuspended in lysis buffer containing 1% Triton X-100, 50 mM Tris HCl (pH 8), 150 mM NaCl, 1% aprotinin-leupeptin, 1 mM Na3VO4, 10 mM NaF, and 1 × Complete (Roche, Mannheim, Germany) for 45 minutes at 4°C. After centrifugation at 15 000 g for 15 minutes at 4°C the supernatant was collected as whole cell lysates. The subcellular fractionation was performed as previously described.29 TOM-1 cells (15 × 106 cells) were lysed in an ice-cold solution containing 5 mM sodium phosphate (pH 7.4), 0.2% Nonidet-P 40, 50 mM NaCl, 150 mM sucrose, 5 mM KCl, 2 mM dithiothreitol, 1 M MgCl2, 0.5 mM CaCl2, 1 mM Na3VO4, 10 mM NaF, and 1 × Complete (Roche). After incubation for 5 minutes, cells were centrifuged at 1000g for 10 minutes at 4°C. The resulting supernatant was collected as the cytoplasmic fraction. The pellet was resuspended in lysis buffer without NP and loaded onto a solution containing 30% (wt/vol) sucrose, 2.5 mM Tris-HCl (pH 7.4), and 10 mM NaCl. After centrifugation at 1000g 10 minutes at 4°C, nuclei were resuspended in an ice-cold solution containing Tris-HCl 50 mM (pH 7.4), 300 mM NaCl, and 0.5% Triton X-100 and incubated in agitation at 4°C for 30 minutes. After centrifugation of the extract at 10 000g for 10 minutes at 4°C, the supernatant was collected as the nuclear fraction. Protein concentration was determined with the BCA assay (Pierce Chemical, West Pico, Rockville, IL).
Western blotting
Equal amounts of protein (15 μg/lane for nuclear and cytoplasmic extract and 50 μg/lane for total extracts) were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto a nitrocellulose membrane (Bio-Rad, Hercules, CA). The membranes were blocked for 1 hour with 0.1% Tropix in 0.01% Tween 20 PBS and incubated with a monoclonal antibody against β-catenin (1:1000 Transduction Laboratories, Lexington, KY) or PARP p85 (1:1000 Promega, Madison, WI). Antibodies against lamin A (1:1000 Transduction Laboratories), β-tubulin, and β-actin (1:4000 Sigma-Aldrich) were used to confirm equal loading of nuclear, cytoplasmic, and total extracts, respectively. Secondary antibodies coupled with alkaline phosphatase (Sigma-Aldrich, 1:10 000) were added for an additional 1 hour. Inmunoreactive bands were developed using a chemiluminescent substrate (CSPD, Tropix) and Hyperfilm ECL plus films (Amersham, Arlington, Heights, IL). An autoradiograph was obtained with exposure times from 1 to 10 minutes.
Treatment with the Wnt inhibitor quercetin
ALL-derived cell lines were grown at a density of 1 × 106 cells/mL in 25-cm2 flasks with 10 mL RPMI 1640 medium supplemented with 20% fetal bovine serum and maintained at 37°C in a humid atmosphere containing 5% CO2. Cells were treated with 50 μM of quercetin (Acros, Geel, Belgium) for 24 hours. After treatment, cells were washed in PBS, pelleted by centrifugation at 1000 g during 5 minutes and frozen at −80°C until use.
Detection of oligonucleosomal fragments
DNA fragmentation was measured using the Cell Death Detection enzyme-linked immunosorbent assay kit (ELISAPLUS; Roche) as recommended by the manufacturer. Briefly, a 96-well microtiter plate was coated with antibody against histones. Diluted samples were then applied to the wells and incubated with peroxidase-conjugated anti-DNA antibody for 120 minutes. Color substrate was added and absorbance was measured at 405 nm using a 96-well microtiter plate reader. The enrichment of mononucleosomes and oligonucleosomes released by cells was calculated as the ratio of the absorbance of treated cells versus absorbance of control cells. This assay has previously been used to detect cell apoptosis and has been demonstrated to correlate with other techniques to detect apoptosis.30-32
Statistical analysis
For statistical purposes, ALL patients were classified into 2 different methylation groups according to the numbers of Wnt-antagonist genes methylated: nonmethylated (no methylated genes) and methylated group (at least one methylated gene). P values for comparisons of continuous variables between groups of patients were 2-tailed and based on the Wilcoxon rank sum test. P values for dichotomous variables were based on the Fisher exact test. The remaining P values were based on the Pearson χ2 test. OS was measured from the day of diagnosis until death from any cause and was censored only for patients known to be alive at last contact. DFS was calculated from the date of first CR until the date of first relapse or the date of death in first CR. Patients alive and still in remission at last follow-up examination were censored in the analysis. Patients who underwent allogeneic SCT were included but censored at the date of transplantation. Distributions of OS and DFS curves were estimated by the method of Kaplan and Meier, with 95% CIs calculated by means of the formula by Greenwood. Comparisons of OS or DFS between groups were based on the log-rank test. Univariate and multivariate analyses (logistic regression model) were used to determine the factors associated with DFS and OS. Stepwise modeling was performed to screen potential variables for inclusion in the final model. Entry criteria for the multivariable Cox regression analysis was a P < .1 in univariate analysis. P values of no more than .05 were taken as the threshold for statistical significance in the final model. All relapse and survival data were updated in March 2006, and all follow-up data were censored at that point.
Results
Expression of Wnt inhibitors in ALL cells is regulated by promoter hypermethylation
We analyzed expression and methylation status of 7 Wnt antagonists (sFRP1, sFRP2, sFRP4, sFRP5, WIF1, DKK3, and HDPR1) in ALL-derived cell lines and lymphoblasts from patients with newly diagnosed ALL. Hypermethylation of the gene promoters of Wnt inhibitors was observed in every cell line (Figure 1A) and was associated with down-regulation of gene expression as demonstrated by restored gene expression after treatment with 5-aza-2′-deoxycytidine in TOM-1 and NALM-20 cell lines (Figure 1B-C). Among 261 ALL patients (Table 1), the methylation frequencies (in descending order) were as follows: 38% for sFRP1, 33% for DKK3, 30% for WIF1, 28% for sFRP5, 26% for HDPR1, 21% for sFRP4, and 16% for sFRP2 (Figure 2A-B). No methylated genes were found in 100 of 261 patients (nonmethylated group, 38%), whereas most ALLs (161 [62%] of 261) had methylation of at least one gene (methylated group), ranging from one to 7 methylated genes (Figure 2C). Other risk factors including cytogenetic groups were similarly distributed between the 2 groups defined by Wnt inhibitors promoter methylation (Table 1). In a small group of patients (n = 17), bone marrow samples were obtained at the time of diagnoses and after relapse for analysis of methylation of Wnt inhibitors. Interestingly, the same genes were methylated at diagnoses and after relapse, suggesting that the methylation pattern is not altered at the time of relapse in the majority of patient (not shown). Samples obtained from patients in CR showed no abnormal methylation of any of the Wnt inhibitors.

Methylation and expression analysis of Wnt-antagonist genes in ALL-derived cell lines. (A) MSP analysis of the methylated sequences in different ALL-cell lines. (B-C) Expression of Wnt inhibitors in TOM-1 (B) and NALM-20 (C) cell lines before and after treatment with the demethylating agent 5-aza-2′-deoxycytidine (Aza) demonstrating an up-regulation of gene expression after treatment. Gene expression was normalized with expression in normal lymphocytes (normalized ratio = 100%). The mean ± SD of 3 different experiments (B-C) is shown.
Methylation and expression analysis of Wnt-antagonist genes in ALL-derived cell lines. (A) MSP analysis of the methylated sequences in different ALL-cell lines. (B-C) Expression of Wnt inhibitors in TOM-1 (B) and NALM-20 (C) cell lines before and after treatment with the demethylating agent 5-aza-2′-deoxycytidine (Aza) demonstrating an up-regulation of gene expression after treatment. Gene expression was normalized with expression in normal lymphocytes (normalized ratio = 100%). The mean ± SD of 3 different experiments (B-C) is shown.

Promoter hypermethylation of Wnt antagonists in samples from patients with ALL. (A) MSP analysis of methylated (M) and unmethylated sequences (U) in patients (UPN) with ALL at diagnosis. PB C− indicates peripheral blood lymphocytes from healthy donors; BM C−, bone marrow mononuclear cells from healthy donors; C+, human male gDNA universally methylated for all genes (used as a positive control for methylated alleles). (B) Frequency of promoter methylation for each Wnt antagonist in a series of 261 patients with ALL at diagnosis. (C) Distribution of patients included in the study according to the number of methylated genes.
Promoter hypermethylation of Wnt antagonists in samples from patients with ALL. (A) MSP analysis of methylated (M) and unmethylated sequences (U) in patients (UPN) with ALL at diagnosis. PB C− indicates peripheral blood lymphocytes from healthy donors; BM C−, bone marrow mononuclear cells from healthy donors; C+, human male gDNA universally methylated for all genes (used as a positive control for methylated alleles). (B) Frequency of promoter methylation for each Wnt antagonist in a series of 261 patients with ALL at diagnosis. (C) Distribution of patients included in the study according to the number of methylated genes.
Hypermethylation of Wnt inhibitors is associated with constitutive activation of the Wnt pathway in ALL
To demonstrate that the Wnt-signaling pathway was activated in ALL, we analyzed the expression of upstream/downstream genes regulated by the Wnt pathway as well as the localization nucleus or cytoplasm of β-catenin.4 Expression of the WNT16, FZ3 receptor, the transcription factors TCF1 and LEF1, and the target gene cyclin D1 was up-regulated in TOM-1 and NALM-20 cell lines in comparison with normal lymphocytes indicating β-catenin–mediated transcriptional activation (Figure 3A-B). In addition, we demonstrated by Western blot that β-catenin was mostly localized in the nuclear fraction (Figure 3C). The fact that treatment with 5-aza-2′-deoxycytidine induced a down-regulation of expression of genes regulated by the Wnt-signaling pathway (Figure 3A-B) and that nuclear localization of β-catenin was reduced (Figure 3C) after demethylation indicates that hypermethylation of Wnt inhibitors is at least in part responsible for constitutive activation of the Wnt pathway in ALL. Consistent with this hypothesis, transcript levels of WNT16 (mean N: 190% versus 90%, P = .006, Figure 4A),FZ3 (mean N: 300% versus 210%, P = .05, Figure 4B), TCF1 (mean N: 260% versus 50%, P = .01, Figure 4C), LEF1 (mean N: 225% versus 25%, P < .001, Figure 4D), and cyclin D1 (mean N: 170% versus 32%, P = .003, Figure 4E) were significantly higher among methylated patients compared with nonmethylated patients.

Methylation of Wnt inhibitors is associated with activation of Wnt-signaling pathway in ALL. (A-B) Expression of Wnt-signaling pathway genes before and after treatment with the demethylating agent 5-aza-2′-deoxycytidine (Aza) in comparison with healthy lymphocytes (normalized ratio = 100%) in TOM-1 (A) and NALM-20 (B) ALL-derived cell lines. The mean ± SD of 3 different experiments is shown. (C) Subcellular location of β-catenin in the ALL cell line TOM-1. β-Catenin is located preferentially in the nucleus indicating activation of the Wnt pathway in ALL. Treatment with 5-aza-2′-deoxycytidine decreases the nuclear location of β-catenin. A representative example of 3 experiments is shown.
Methylation of Wnt inhibitors is associated with activation of Wnt-signaling pathway in ALL. (A-B) Expression of Wnt-signaling pathway genes before and after treatment with the demethylating agent 5-aza-2′-deoxycytidine (Aza) in comparison with healthy lymphocytes (normalized ratio = 100%) in TOM-1 (A) and NALM-20 (B) ALL-derived cell lines. The mean ± SD of 3 different experiments is shown. (C) Subcellular location of β-catenin in the ALL cell line TOM-1. β-Catenin is located preferentially in the nucleus indicating activation of the Wnt pathway in ALL. Treatment with 5-aza-2′-deoxycytidine decreases the nuclear location of β-catenin. A representative example of 3 experiments is shown.

Hypermethylation of Wnt inhibitors is associated with up-regulation of genes implicated in the Wnt-signaling pathway in ALL patients. Expression of the Wnt pathway genes in ALL patients was measured by qRT-PCR. Significantly higher levels of WNT16 (A), FZ3 (B), TCF1 (C), LEF1 (D), and cyclin D1 (E) transcripts were detected among methylated ALL patients compared with unmethylated patients. Bars represent the mean expression (95% CI) in patients with ALL in comparison with healthy lymphocytes (normalized ratio = 100%).
Hypermethylation of Wnt inhibitors is associated with up-regulation of genes implicated in the Wnt-signaling pathway in ALL patients. Expression of the Wnt pathway genes in ALL patients was measured by qRT-PCR. Significantly higher levels of WNT16 (A), FZ3 (B), TCF1 (C), LEF1 (D), and cyclin D1 (E) transcripts were detected among methylated ALL patients compared with unmethylated patients. Bars represent the mean expression (95% CI) in patients with ALL in comparison with healthy lymphocytes (normalized ratio = 100%).
Inhibition of Wnt signaling leads to cell apoptosis in ALL
To determine whether constitutive activation of the Wnt signaling pathway is implicated in the pathogenesis of ALL, TOM-1, and NALM-20 cell lines were treated with quercetin, an inhibitor of β-catenin/Tcf transcriptional activity.33 Incubation of ALL cells with quercetin for 24 hours induced down-regulation of TCF1, LEF1, and cyclin D1 mRNA expression (Figure 5A-B) and increased apoptosis as detected by an increase in oligonucleosomal fragments (Figure 5C) and confirmed by Western blot analysis against PARP fragment (Figure 5D).

Inhibition of the Wnt signaling by quercetin induces down-regulation of Wnt target genes and apoptosis of ALL cells. Expression of Wnt-signaling pathway genes before and after treatment with quercetin in TOM-1 (A) and NALM-20 (B) cell lines. Expression was normalized in comparison with healthy lymphocytes (Normalized ratio = 100%). The mean ± SD of 3 different experiments is shown. (C) Apoptosis of ALL cells was significantly higher after treatment with quercetin (P < .05). (D) Expression of PARP was increased after treatment with 5-aza-2′-deoxycytidine (Aza) and quercetin (Quer). A representative example of 3 experiments is shown.
Inhibition of the Wnt signaling by quercetin induces down-regulation of Wnt target genes and apoptosis of ALL cells. Expression of Wnt-signaling pathway genes before and after treatment with quercetin in TOM-1 (A) and NALM-20 (B) cell lines. Expression was normalized in comparison with healthy lymphocytes (Normalized ratio = 100%). The mean ± SD of 3 different experiments is shown. (C) Apoptosis of ALL cells was significantly higher after treatment with quercetin (P < .05). (D) Expression of PARP was increased after treatment with 5-aza-2′-deoxycytidine (Aza) and quercetin (Quer). A representative example of 3 experiments is shown.
Hypermethylation and silencing of Wnt inhibitors is associated with poor prognosis in ALL
Table 1 details the relapse history, CR rates, and mortality for patients included in the different methylation groups. CR rates of patients in the methylated and nonmethylated groups were 93% and 87%, respectively, accounting for an overall CR rate of 90%. This suggests that Wnt methylation profile did not correlate with response to remission induction therapy. However, patients in the nonmethylated group had a lower relapse rate than patients in the methylated group (29% versus 59%, P < .001). Mortality rate was also lower for nonmethylated group compared with methylated group (36% versus 59%, P = .001). Similar results were obtained in a separate analyses of children (relapse rate, 19% for nonmethylated group versus 47% for methylated group, P = .002; mortality rate, 16% for nonmethylated group versus 30% for methylated group, P = .05) and adults (relapse rate, 43% for nonmethylated group versus 72% for methylated group, P = .006; mortality rate, 62% for nonmethylated group versus 83% for methylated group, P = .01).
Estimated rates of DFS at 10 years were 66% and 25% for nonmethylated and methylated groups, respectively (P < .001; Figure 6A). Among children, the 10-year DFS was 77% for the nonmethylated group and 34% for methylated patients (P < .003; Figure 6B), whereas in adults with ALL, the 10-year DFS was 48% for the nonmethylated group and 15% for methylated group (P = .005; Figure 6C). The actuarial OS at 10 years calculated for all leukemic patients was 61% for nonmethylated patients and 28% for methylated patients (P < .001). Significant differences were observed in the actuarial OS among nonmethylated and methylated groups in the separate analyses of children (82% and 49%, respectively, P = .05) and adults (34% and 10%, respectively, P = .01).

Kaplan-Meier survivor function for patients with ALL. DFS curves for all the patients enrolled in this study (A), childhood ALL (B), and adult ALL (C) according to the methylation profile. Solid lines indicate nonmethylated patients; dashed lines, methylated patients.
Kaplan-Meier survivor function for patients with ALL. DFS curves for all the patients enrolled in this study (A), childhood ALL (B), and adult ALL (C) according to the methylation profile. Solid lines indicate nonmethylated patients; dashed lines, methylated patients.
A multivariate analysis of potential prognostic factors demonstrated that hypermethylation profile was an independent prognostic factor in predicting DFS in the global series (P<.001) as well as in both childhood (P = .001) and adult ALL (P = .004; Table 2). Methylation status was also independently associated with OS in the global series (P = .01), adult ALL (P = .04) and childhood ALL (P = .01).
Multivariate Cox model for DFS
| Feature | Univariate analysis, P | Multivariate analysis, P |
|---|---|---|
| Global series | ||
| WNT methylation | < .001 | < .001 |
| WBC count greater than 50 × 109/L | .006 | .05 |
| BCR-ABL positivity | < .001 | < .001 |
| T phenotype | .05 | NS |
| Age older than 15 y | .001 | .003 |
| PETHEMA poor risk | .05 | NS |
| Childhood ALL | ||
| WNT methylation | < .001 | .001 |
| NCI poor risk | .04 | NS |
| T phenotype | .04 | NS |
| WBC count greater than 50 × 109/L | .03 | NS |
| Adult ALL | ||
| WNT methylation | .005 | .004 |
| T phenotype | .05 | NS |
| WBC count greater than 50 × 109/L | .05 | NS |
| BCR-ABL positivity | <.001 | .001 |
| Feature | Univariate analysis, P | Multivariate analysis, P |
|---|---|---|
| Global series | ||
| WNT methylation | < .001 | < .001 |
| WBC count greater than 50 × 109/L | .006 | .05 |
| BCR-ABL positivity | < .001 | < .001 |
| T phenotype | .05 | NS |
| Age older than 15 y | .001 | .003 |
| PETHEMA poor risk | .05 | NS |
| Childhood ALL | ||
| WNT methylation | < .001 | .001 |
| NCI poor risk | .04 | NS |
| T phenotype | .04 | NS |
| WBC count greater than 50 × 109/L | .03 | NS |
| Adult ALL | ||
| WNT methylation | .005 | .004 |
| T phenotype | .05 | NS |
| WBC count greater than 50 × 109/L | .05 | NS |
| BCR-ABL positivity | <.001 | .001 |
Discussion
Abnormal Wnt signaling has become a hallmark in a number of solid tumors like colon cancer where mutations mainly in the APC genes, but also in other components of the “destruction complex,” result in constitutive activation of β-catenin and malignant transformation.1,11 Recent findings regarding the role of the Wnt-signaling pathway in T- and B-lymphocyte development as well as in self-renewal of stem cells have also suggested that alterations in this pathway are likely implicated in leukemogenesis.4,18,34 Although a role for activation of the Wnt-signaling pathway in the pathogenesis of acute myeloid leukemia,34 chronic lymphocytic leukemia,18 chronic myeloid leukemia,35 or multiple myeloma36 has been demonstrated, no mutations of Wnt-signaling genes have been detected suggesting that abnormal regulation of this pathway is mediated by other mechanisms.
The existence of an autocrine loop that activates β-catenin by up-regulation of Wnt signaling molecules has been demonstrated in myeloid and lymphoid malignancies. For instance, in pre-B-cell leukemia, the E2A-PBX1 translocation has been shown to up-regulate expression of Wnt16 contributing to the development of the disease,37 whereas in multiple myeloma, other Wnt molecules (Wnt5a, Wnt10b, or Wnt16) have been shown to be up-regulated and associated with β-catenin activation and nuclear localization.36 Other mechanisms of activation have been described in patients with acute myeloid leukemia where commonly observed translocations such as AML1-ETO, PML-RARA, or PLZF-RARA induced increased expression of plakoglobin, which in turn might lead to increased Wnt signaling similar to the effects of increased levels of β-catenin.38
The findings described in our study demonstrate that epigenetic regulation of Wnt inhibitors is another mechanism that significantly contributes to activation of the Wnt-signaling in ALL. Although, hypermethylation of Wnt inhibitors has been recently described in patients with solid tumors,17,39-41 CLL,13 or even promyelocytic leukemia,42 our work demonstrates for the first time the relation between hypermethylation of Wnt inhibitors and activation of the Wnt signaling pathway in ALL. The nuclear localization of β-catenin and up-regulation of Wnt target genes in ALL cells, specifically TCF1, LEF1, direct targets of β-catenin–mediated transcriptional activation,43 as well as down-regulation of their expression and decreased in nuclear β-catenin induced by demethylation of Wnt inhibitors with 5-aza-2′-deoxycytidine clearly indicates that epigenetic regulation is responsible at least in part for activation of the Wnt signaling in ALL. We did not examine protein expression of Wnt inhibitors in part due to the fact that the goal of our study was to demonstrate that regulation expression of Wnt inhibitors was at least in part due to epigenetic mechanisms in patients with ALL and also because the activation of the Wnt pathway is demonstrated by the nuclear location of β-catenin and the reduced nuclear location after 5-aza-2′-deoxycytidine treatment.
Recent studies have suggested that Wnt5a besides activating the canonical Wnt signaling may signal through the noncanonical Wnt/Ca++ pathway to suppress cyclin D1 expression inhibiting B-cell proliferation and acting as a tumor suppressor gene.44 Furthermore, expression of WNT5A was down-regulated in the majority of patients with acute leukemia.44 In our study, baseline expression of cyclin D1 in methylated patients was significantly higher than in normal lymphocytes (Figure 4) and was only down-regulated after treatment with either the demethylating agent 5-aza-2′-deoxycytidine or after inhibition of β-catenin with quercetin. Although we did not examine expression of WNT5A, these results suggest a role of the canonical Wnt-signaling pathway in regulation of cyclin D1 in patients with ALL.
We have previously demonstrated 20-22,45-49 that methylation in human ALL cells can participate in the inactivation of 3 key cellular pathways: (1) growth-deregulating events comprising those that target the principal late G1 cell cycle checkpoint either directly (P15, P16, and P57 inactivation) or indirectly (P73, PTEN, NES1, and LATS2 inactivation) and those that regulate the G2/M transition down-regulating CDC2/cyclin A kinase activity (LATS-1); (2) the apoptotic program through inactivation of P14, TMS1, APAF1, ASPP1, DIABLO, and DAPK; and (3) the cell-to-cell adhesion by the inactivation of some members of the cadherin (CDH13 and CDH1) and metalloproteinase (ADAMTS1 and ADAMTS5) families. Moreover, aberrant methylation of CpG islands is quantitatively different in individual tumors within the same tumor type, and this patient-specific methylation profile provides important prognostic information in patients with ALL. Similarly, in the current study we divided the methylation groups into nonmethylated (no methylated genes) and methylated group (at least one methylated gene) as the statistical analyses of relapse rate, mortality rate, DFS, and OS according to the number of methylated genes (from 0 to 7) showed that there was no statistically significant difference in prognosis between patients with one to 7 methylated genes, but there was a large and significant difference in comparison with nonmethylated patients. The presence in individual tumors of multiple epigenetic events that affect each of the pathways discussed is a factor of poor prognosis in ALL. Our results not only provide further support for this hypothesis but also establish a potential mechanism by which hypermethylation contributes to the pathogenesis of ALL.
The current study also has potential clinical implications for patients with ALL. We clearly demonstrate that the methylation phenotype has a prognostic value both in terms of DFS and OS. Hence, the analysis of promoter methylation in patients with ALL could contribute to tailoring treatment strategies according to the prognosis. Most importantly, the demonstration that inhibition of β-catenin induces apoptosis of ALL establishes this pathway as an attractive target for the use of more specific therapies such as quercetin33 or more recently developed pharmacologic antagonists of the oncogenic Tcf/β-catenin protein complex.50 Furthermore, the fact that treatment with 5-aza-2′-deoxycytidine inactivates the Wnt-signaling pathway, as demonstrated in this study, may warrant the use of clinically approved demethylating agents like decitabine or 5-aza-2′-deoxycytidine in patients with ALL.51
Authorship
Contribution: J.R.G., F.P., and X.A. performed and designed research, collected and analyzed data, and wrote the manuscript; L.C., A.J.V., E.S.J.E., and L.G. performed research and collected data; and M.J.C., A.T., and A.H. interpreted data and approved the final version of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: José Román-Gómez, Hematology Department, Reina Sofia Hospital, Avda Menendez Pidal s/n, 14004 Cordoba, Spain; e-mail: peperosa@teleline.es; and Felipe Prosper, Hematology and Cell Therapy, Clínica Universitaria, Avda, Pío XII 36, Pamplona 31008, Spain; e-mail: fprosper@unav.es.
An Inside Blood analysis of this article appears at the front of this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported in part by grants from Fondo de Investigaciones Sanitarias PI030661, PI030141, PI060003; Junta de Andalucía 03/143, 03/144; RETIC C03/10; Fundación de Investigación Médica Mutua Madrileña Automovilista, Fundación la Caixa (001); Fundación IMABIS; and Asociacion Medicina e Investigacion (AMI). This project was funded through the “UTE project CIMA.”

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal