Abstract
Seventy-seven potential analogs of phospholipid polar heads, choline and ethanolamine, were evaluated in vitro as inhibitors ofPlasmodium falciparum growth. Their IC50 ranged from 10−3 to 10−7 mol/L. Ten compounds showed similar antimalarial activity when tested against three different parasite strains (2 chloroquine-sensitive strains and 1 chloroquine-resistant strain). Compounds showing marked antimalarial activity were assayed for their effects on phospholipid metabolism. The most active compounds (IC50 of 1 to 0.03 μmol/L) were inhibitors of de novo phosphatidylcholine (PC) biosynthesis from choline. For a series of 50 compounds, there was a close correlation between impairment of phospholipid biosynthesis and inhibition of in vitro malaria parasite growth. High choline concentrations caused a marked specific shift in the curves for PC biosynthesis inhibition. Concentrations inhibiting 50% PC metabolism from choline were in close agreement with the Ki of these compounds for the choline transporter inPlasmodium knowlesi-infected erythrocytes. By contrast, measurement of the effects of 12 of these compounds on rapidly dividing lymphoblastoid cells showed a total absence of correlation between parasite growth inhibition and human lymphoblastoid cell growth inhibition. Specific antimalarial effects of choline or ethanolamine analogs are thus likely mediated by their alteration of phospholipid metabolism. This indicates that de novo PC biosynthesis from choline is a very realistic target for new malaria chemotherapy, even against pharmacoresistant strains.
THE INCREASING polypharmacoresistance ofPlasmodium falciparum to conventional antimalarials, combined with the resistance of mosquitoes to various pesticides, has contributed to a dramatic resurgence of malaria.1 New therapeutic approaches to this endemic disease are now being actively investigated. One approach consists of interfering with parasite metabolic pathways to develop a new range of antimalarial drugs with original structures and modes of action.
We previously characterized phospholipid (PL) metabolism as an ideal target for new chemotherapy due to its vital importance to the parasite. PL metabolism is absent from normal mature human erythrocytes,2 but the erythrocyte PL content increases by as much as 500% after infection.3-5 Phosphatidylcholine (PC) and phosphatidylethanolamine (PE) are the major PL of the infected erythrocyte, representing about 85% of total PL. De novo pathways for PC and PE biosynthesis from choline and ethanolamine, respectively, have been thoroughly characterized in Plasmodium-infected erythrocytes.6 For de novo PC biosynthesis, cholinephosphate cytidylyltransferase is a regulatory step in the pathway, but choline transport (which regulates the supply of precursor) is also a limiting step.7 For de novo PE biosynthesis, ethanolaminephosphate cytidylyltransferase is probably the rate-limiting step (personal observation), whereas ethanolamine entry occurs by mere passive diffusion.8 We have previously shown that impairment of PL biosynthesis with polar head analogs, which interfere with natural polar head incorporation either by substitution or competition9-11 or with unnatural fatty acids,12 is lethal to the intraerythrocytic stage ofP falciparum in vitro.
We have just reported the effects of 77 compounds that are analogs of ethanolamine or, for most of them, of choline on in vitro P falciparum growth.13 In the present study, systematic screening of compounds with antimalarial effects on PL metabolism, using a method that we developed,14 enabled us to show a clear correlation between their antimalarial activity and PL metabolism impairment. Some pharmacologic target characteristics that will be useful for designing very active specific antimalarial compounds could thus now be defined.
MATERIALS AND METHODS
Chemicals.
Thirty products were chemically synthesized as described previously.13 The compounds that were synthesized for this study are noted in Table 1. F14 and F19 were provided by Dr J. Berthe (Sanofi, Montpellier, France). The other commercial products, as well as choline, ethanolamine, and dibutyl phtalate, came from Sigma Chemical Co (St Louis, MO).
(Methyl-3H)choline, (1-3H)ethan-1-ol-2-amine, and (G-3H)hypoxanthine were purchased from Amersham Corp (Les Ulis, France) and (3H)thymidine was purchased from CEA (Saclay, France). RPMI 1640 medium15 and special RPMI 1640 without choline, methionine, or serine were obtained from GIBCO Laboratories (Eragny, France). Modified RPMI 1640 consisted of special RPMI 1640 complemented with 25 mmol/L HEPES, 20 μmol/L methionine, and 50 μmol/L serine. All reagents were of analytical grade.
Biologic materials.
Human blood or AB human serum came from the local blood bank. ThreeP falciparum strains were routinely used: 2 chloroquine-sensitive strains, the Nigerian strain16 and the NF54 strain,17 and a chloroquine-resistant strain from Thailand (T23).18P falciparum strains were maintained by serial passages in human erythrocytes according to the petri dish candle-jar method.19P knowlesi-infected erythrocytes (Washington strain, variant 1) were collected from splenectomized Macaca fascicularis monkeys (Sanofi, Montpellier, France) infected with cryopreserved parasites, as previously described.9 The lymphoblastoid cell line (SAR strain) was a gift from Dr B. Klein (Institut de Génétique Moléculaire, Montpellier, France).
Antimalarial activity.
Effects of drugs on in vitro P falciparum growth were measured in microtiter plates using (3H)hypoxanthine incorporation into nucleic acids of an infected erythrocyte suspension (final hematocrit level, 1%; parasitemia, 0.3% to 0.8%) according to the method of Desjardins et al,20 with the drug placed in contact with infected erythrocytes for one full parasite cycle (48 hours). Parasite viability was assayed by the capacity to incorporate (3H)hypoxanthine (0.7 μCi/well) in parasite nucleic acids for 15 hours. At the end of incubation, cells were collected on glass fiber filters (Whatman GF/C; Whatman, Maidstone, UK) with a cell harvester (Micro 96; Skatron, Lier, Norway) and filters were then counted for radioactivity in 2 mL of scintillation cocktail (Packard no. 299) (Packard Instrument, Meriden, CT) in a Beckman LS 5000 spectrophotometer (Beckman, Fullerton, CA). The results are expressed as the concentration resulting in 50% inhibition (IC50) of hypoxanthine incorporation. Experiments were performed at least twice in triplicate.
Measurement of PL metabolism.
PL metabolism was monitored in microtiter plates by the incorporation of 10 μmol/L (3H)choline (1.75 Ci/mmol) or 2 μmol/L (3H)ethanolamine (2.9 Ci/mmol) into PL constituents as previously described.14 For comparison and to determine any specific effects, incorporation of (3H)hypoxanthine (1.2 μCi/well, 6 Ci/mmol) into nucleic acids was monitored concurrently. Unless otherwise specified, infected erythrocyte suspensions (30 μL, 1 to 5 × 106 infected cells/well, 3% to 10% parasitemia) were mixed with 20 μL of modified RPMI 1640 without (control) or with the drug. In some experiments, incubations were performed at a lower hematocrit level, using twofold less cells in a final volume of 300 μL. After 5 minutes at 37°C, radioactive precursors were added (10 μL) either for 1 hour (short period) or 4 hours (long period) using the candle-jar method.19 After incubation, cells were collected on glass fiber filters (Whatman GF/C) using a cell harvester and lysed with water. Insoluble material (such as nucleic acids, proteins, and lipids) retained on the filter was abundantly washed with water (for 50 seconds). Filters were then dried for 35 seconds and placed in scintillation vials containing 2 mL of scintillation cocktail. Final counting was performed after 24 hours at room temperature. Blank values were obtained by incubating an equal number of noninfected erythrocytes. When choline was used as radioactive precursor, filters were presoaked in 0.05% polyethyleneimine to eliminate nonspecific binding to the filter. Blank values were deduced from the total radioactivity incorporated by infected cells.
Measurement of choline transport inhibition.
Pure P knowlesi-infected erythrocytes were isolated after Percoll/sorbitol fractionation according to Kutner et al21and then abundantly washed with special RPMI before choline influx measurement, as described previously.8 Briefly, pure infected erythrocytes (5 to 7 × 107 cells/100 μL, trophozoites) were preincubated for a short period (5 to 10 minutes) at 37°C in the presence of 750 μL modified RPMI containing the drug or not containing the drug (control). Choline influx was measured after the rapid addition of 50 μL (3H)choline (specific activity, 0.2 Ci/mmol) to give a final hematocrit level of 0.6% to 0.8%. After 6 minutes of incubation at 37°C, the flux was stopped by adding 2.5 mL of ice-cold modified RPMI. Triplicate 1-mL portions of cell suspension were immediately overlaid on 400 μL of ice-cold n-dibutyl phtalate (density, 1.04 g/mL) in polyethylene tubes and centrifuged in a Beckman 11 Microfuge at 10,000g for 10 seconds at 4°C. Supernatants were discarded, and 2.5 mL of cold special RPMI was carefully added on the dibutyl phtalate layer to wash the walls of the microtubes. After centrifugation, supernatants and dibutyl phtalate were discarded and cells were lysed and precipitated with 500 μL of 10% (wt/vol) trichloracetic acid. After centrifugation, 400 μL of the supernatant was counted for radioactivity in 10 mL scintillation cocktail.8 Nonspecific choline transport was determined under the same conditions but in the presence of 1.2 mmol/L of choline in the incubation medium.
Measurement of toxicity against a lymphoblastoid cell line (SAR strain).
Cells were routinely cultured at 37°C in RPMI 1640 medium complemented with 50 μmol/L β mercaptoethanol, 1 mmol/L glutamine, and 10% fetal calf serum (GIBCO). The effect of drugs on cell viability was measured in microtiter plates after (3H)thymidine incorporation into nucleic acids of the lymphoblastoid cell suspension (4,500 cells/well). Cells were first exposed to various drug concentrations for 24 hours (1 cell cycle) at 37°C and then (3H)thymidine (0.75 μCi/well) was added for an additional 5 to 6 hours. At the end of incubation, cells were harvested on glass fiber filters as described above, and filters were then counted for radioactivity in 2 mL scintillation cocktail. The results are expressed as the concentration resulting in 50% inhibition (LV50) of thymidine incorporation.
RESULTS
Drug effect on P falciparum growth in vitro.
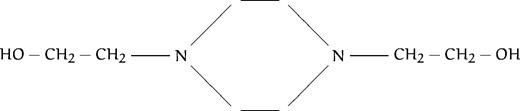
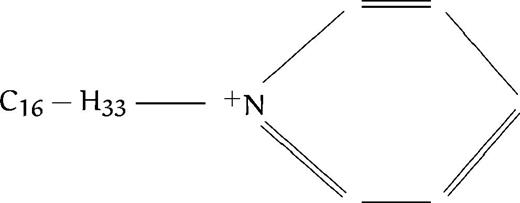
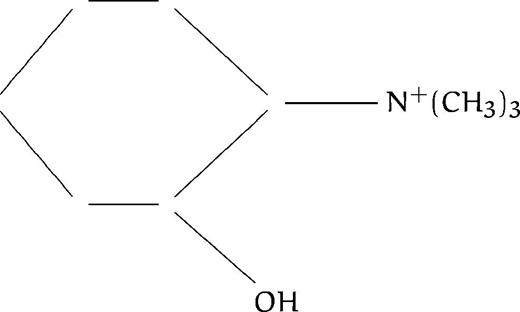
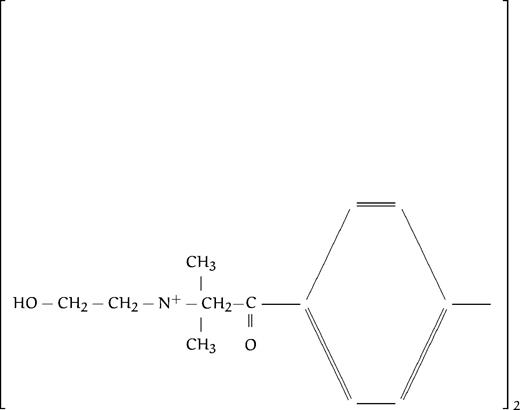
We have just reported the in vitro antimalarial activity of 77 compounds, analogs to ethanolamine or choline (listed in Table 1), along with structure activity relationships, to define the general structural features involved in the activity so as to be able to design highly efficient antimalarial compounds.13 All compounds contained at least one amino group (substituted or not), as in the ethanolamine or choline molecule. We distinguished eight different groups based on the degree of substitution of the amine function: group A corresponded to compounds with primary amines; group B with secondary amines, group C with tertiary amines; and groups D, E, F, G, and H with quaternary ammonium groups (the last 4 series are described in more detail below). The present study was aimed at thoroughly studying the mechanism of action of these compounds to draw up the pharmacologic target characteristics. Regarding the antimalarial activity, results reported in our previous study showed that all of the compounds were lethal to P falciparum in vitro in a dose-dependent manner, with IC50 ranging from 4.8 mmol/L to 0.033 μmol/L (Table1). Amine compounds in groups A, B, and C (except N-substituted compounds with a long alkyl chain, see Discussion) exhibited far lower antimalarial activities (IC50 ranging from 10−3 to 10−5 mol/L) than compounds containing one (group E or F) or two (group G, H) quaternary ammonium groups, with IC50 ranging from 10−4 to 10−7 mol/L. The most active compounds were quaternary mono- or bis-ammonium salts with small polar head groups, eg, trimethyl, dimethyl (or diethyl) hydroxyethyl, triethyl or tripropyl, N substituted analogs or methyl pyrrolidinium, possessing a long lipophilic alkyl chain constituted of at least eight methylene groups (see Calas et al13 and Discussion).
Ten compounds belonging to groups A, E, F, and G (noted in Table 1) were then tested both against another chloroquine-sensitive strain (NF54) and one chloroquine-resistant strain (T23; IC50against chloroquine = 0.03 μmol/L and 0.6 μmol/L, respectively; personal observation, 1996), in addition to the Nigerian strain (IC50 = 0.02 μmol/L; personal observation, 1996) that was routinely used in this study. Regardless of the compound and the group to which it belonged, the IC50 against the three strains were very similar, differing by less than threefold (these values are thus not reported in Table 1). These compounds could thus also be efficient against pharmacoresistant strains.
Drug effect on macromolecule biosyntheses.
The effect of 56 of the most active compounds on PC and PE biosynthesis was evaluated by measuring the incorporation of radioactive choline into PC as well as the incorporation of radioactive ethanolamine into PE using a cell harvester for rapid serial determination.14Incubations were performed for 4 hours or less (1 hour) to determine an early effect of the compounds. We also measured the effects of the compounds on the incorporation of radioactive hypoxanthine* to determine any possible specificity toward PL metabolism versus biosynthesis of other macromolecules such as nucleic acids. The results were expressed as PL50 and NA50 (corresponding to the drug concentration that reduced the amount of synthesized PL or nucleic acids by 50%, respectively).
Figure 1 shows the typical sigmoid dose-response curves obtained with dimethyl-n-pentyl (2-hydroxyethyl) ammonium (F2) when tested on choline, ethanolamine, and hypoxanthine incorporation into macromolecules. This compound inhibited PE and nucleic acid biosynthesis at very high concentrations (NA50, 2.6 mmol/L; PE50, ≥20 mmol/L), which probably corresponded to an alteration of parasite viability. By contrast, it specifically inhibited choline incorporation into PC at much lower concentrations (PC50, 32 μmol/L) and the PC50 was very close to the IC50 (81.3 μmol/L). Fifty-five other compounds were also tested for their effects on macromolecule biosynthesis. Their PL50 and NA50 are listed in Table 1. It should be noted that all group A compounds (containing a primary amine) that specifically impaired the metabolism of ethanolamine (except A7, which impaired choline metabolism) required longer incubation (>1.5 hours) to exert their effects. We have previously shown that A6, which has a free hydroxyethyl group, was incorporated by the parasite into an unnatural PL, ie, phosphatidyl-2-amino-1-butanol, that accumulated at the expense of PE.9 The need to accumulate sufficient amounts of false PL could explain the relatively long time required for these compounds to exert their effects. In contrast, specific compounds containing a tertiary amine or quaternary ammonium (groups C, E, F, G, and H) impaired choline incorporation into PC as early as 15 to 30 minutes (Ancelin and Vial11 and data not shown), probably due to choline transport inhibition (see below). PL50 and NA50 values given in Table 1 correspond to values obtained after 4 hours of incubation, ie, the time needed for both types of analogs to exert a substantial effect.

Effect of F2 on radioactive choline, ethanolamine, and hypoxanthine incorporations into the macromolecules of P falciparum-infected erythrocytes. Infected cells (10% hematocrit, 3.6% parasitemia) were incubated for 4 hours at 37°C in the absence (control) or presence of the indicated concentrations of F2 and in the presence of 10 μmol/L (3H)choline (1.2 μCi) (•), 2 μmol/L (3H)ethanolamine (0.3 μCi) (▴), and trace concentrations of (3H)hypoxanthine (1 μCi) (▪). The reaction was stopped by cell filtration as described in the Materials and Methods. Results are expressed as a percentage of the control (without drug) ± SEM.
Effect of F2 on radioactive choline, ethanolamine, and hypoxanthine incorporations into the macromolecules of P falciparum-infected erythrocytes. Infected cells (10% hematocrit, 3.6% parasitemia) were incubated for 4 hours at 37°C in the absence (control) or presence of the indicated concentrations of F2 and in the presence of 10 μmol/L (3H)choline (1.2 μCi) (•), 2 μmol/L (3H)ethanolamine (0.3 μCi) (▴), and trace concentrations of (3H)hypoxanthine (1 μCi) (▪). The reaction was stopped by cell filtration as described in the Materials and Methods. Results are expressed as a percentage of the control (without drug) ± SEM.
In group A, most compounds except A9, ie, cadaverin, a putrescine homologue, and A2, a sulfhydryl compound, with a wide variety of biologic effects22 appeared to specifically act on the biosynthesis of one PL, whereas biosyntheses of the other PL and of nucleic acids were affected at much higher drug concentrations. They generally acted on ethanolamine incorporation, except for A7, which was more PC-specific. In addition, their PL50 were generally in the same range as their IC50.**This was not the case for A5, which can be considered as a serine analog probably acting on serine metabolism (as an amino acid or PL polar head analog).
The few compounds in group B, regardless of their activity, were nonspecific with respect to PE and PC metabolism. Furthermore, most PL50 and NA50 values differed markedly from the IC50 and there was no correlation between the effects on PL (or nucleic acid) metabolism and the effects on parasite growth. This was especially true for N-substituted compounds with a long alkyl chain (12 carbon atoms, B3 and B4; see below). It should be noted that very few compounds were tested in this series, making it difficult to draw any conclusions for this group.
Concerning group C, PL-specific compounds that showed good PL50/IC50 correlations were the only ones in which N was not included in a cyclic structure and was substituted with a short alkyl chain (C5, C6, and C7). As in group B, when C8 was N-substituted by a long alkyl chain (12 carbon atoms), its action was neither PL-specific nor growth-correlated. When nitrogen was included in the ring structure (C1, C2, C3, and C4), the effects were dependent on the number of atoms in the heterocycle. The actions of molecules with a 6-atom heterocycle (C3 and C4) were not PL-specific or growth-correlated; C2, which has a ring of four carbon atoms, was PL-specific and its PE50 was more correlated with its IC50 than with the PC50, which was lower. Lastly, C1, which contained an aziridine group, acted on PL metabolism but also acted on nucleic acid synthesis at lower concentrations.
Compounds of group E (quaternary ammonium salts with 4 alkyl chains) were specific to PC metabolism provided that they were N-substituted with an alkyl chain having less than 12 carbon atoms (E1, E2, E3, E4, and E5). With longer alkyl chains (12, 14, or 16 atoms, ie, E6, E7, E10, E13, E20, E40, E8, and E50), the compounds acted almost identically and at similar concentrations on PC and nucleic acid metabolism. We have previously shown that compounds such as E4, E6, E7, or E8 acted very quickly, ie, as early as 30 minutes, but the earliest effect was on PL metabolism.10 After 1 hour of incubation, the drug can thus also affect nucleic acid metabolism, which may explain the absence of any differential effect on PL and nucleic acids for these very rapid acting drugs. Nevertheless, the long alkyl chain also seems to be responsible for the absence of correlation with the IC50.
In group F (quaternary ammonium with 3 alkyl and 1 hydroxyethyl N-substitution), we observed the same pattern of effects according to the length of the alkyl chain. Indeed, N-substituted compounds with short alkyl chains (up to 5 atoms, F1 and F2) were PC-specific and their effect on PC metabolism was also correlated with parasite growth. The presence in the short alkyl chain of a ring structure with six carbon atoms (aromatic [F12, F13, and F14] or nonaromatic [F15 and F16]) also led to compounds that were very PC-specific, with PL50 highly correlated with the IC50. Compounds with 10 and 12 carbon atoms showed PC-specific inhibition, but this was not correlated with growth inhibition and increasing the length of the alkyl chain (up to 18 carbon atoms) produced compounds that were neither PC-specific nor correlated (F5, F6, F9, and F10), as already shown for group E compounds, and this was also the case when two long alkyl chains were present as N substitutions (F7 and F11).
All compounds of group G and H with two quaternary ammonium groups were tested for their effect on PL metabolism. Except for H0, which was only slightly more specific to PL (especially PE) than to nucleic acid metabolism, all acted very specifically on de novo PC biosynthesis, regardless of the distance between the two nitrogens, from 5 to 12 methylene groups. In this case also, a short alkyl chain between the two nitrogens resulted in a high PL50-IC50correlation (see G1 or G20), whereas for 10 to 12 methylenes, the compounds showed a slightly lower PL50-IC50correlation (see G3, G4, G23, or H3). On the other hand, HC3 (whose alkyl chain between the two N contained two aromatic rings) was PC-specific and showed a high PL50-IC50correlation.
Effect of hematocrit level.
The relatively high number of compounds with long alkyl chains in amine groups (B or C) and quaternary ammonium salts (group E, F, or G) that showed no PL50/IC50 correlation prompted us to investigate whether the difference between the hematocrit level used in the growth inhibition assay (0.5% to 1.5%) and the hematocrit level used in the PL inhibition assay (8% to 20%) could account for the difference between IC50 and PL50. For some compounds, notably chloroquine, such a hematocrit-dependent effect of the IC50 has already been reported.23 We thus compared the effects of several drugs on macromolecular biosyntheses (PL and nucleic acids) at two different hematocrit levels, generally differing by 10-fold. Figure 2 shows that, with tetradecyltrimethyl ammonium (E7), a 10-fold increase in the hematocrit level led to a 5- to 10-fold increase in the PL50 and in the NA50. It is particularly interesting that, at a low hematocrit level (ie, 1.3%), the PC50 (1.8 μmol/L) was in close agreement with the IC50 (0.9 μmol/L). Twelve other compounds were tested on PL metabolism at two hematocrit levels (Table 2). The same hematocrit level effect was observed with E40 and E8, compounds containing 14 and 16 carbon atoms, respectively. This was also the case for F5, F9, and F6, with 14, 14, and 18 carbon atoms, respectively. By contrast, for compounds with an alkyl chain containing less than 12 carbon atoms, regardless of the group to which they belonged, ie, E, F or G (see E4, F3, or G3), hematocrit level variations caused no marked difference in the PL50. Compounds with 12 carbon atoms had intermediary results, depending on the group and thus on nitrogen substitution (secondary amine, N-trialkyl or N-dialkyl hydroxyethyl monoquaternary ammonium, or bis quaternary ammonium compounds): B4 showed a marked hematocrit level effect, E6 and F4 showed a less pronounced effect (2.5- to 4-fold), and no effect was observed with G4. By contrast, decreasing the hematocrit level did not modify the specificity pattern, regardless of the group and the methylene number of the long hydrophobic alkyl chain.

Effect of hematocrit level on the inhibition of PL and nucleic acid biosyntheses by E7. Experiments were performed for 4 hours at 37°C with the same infected cell suspensions (5.6% parasitemia) either under the high hematocrit level (13%) conditions described in the Materials and Methods, ie, in 60 μL final volume (5 × 106 parasites) (solid symbols) or at low hematocrit level (1.3%) in a final volume of 300 μL (containing 2.5 × 106 parasites) (open symbols), resulting in a 10-fold difference in hematocrit level. In both cases, the concentrations and the specific activities of the radioactive precursors were similar, ie, 10 μmol/L (3H)choline at 1.92 Ci/mmol, 2 μmol/L (3H)ethanolamine at 2.74 Ci/mmol, and trace concentration of (3H)hypoxanthine at 4 μCi/well. The results are derived from a typical experiment performed in triplicate.
Effect of hematocrit level on the inhibition of PL and nucleic acid biosyntheses by E7. Experiments were performed for 4 hours at 37°C with the same infected cell suspensions (5.6% parasitemia) either under the high hematocrit level (13%) conditions described in the Materials and Methods, ie, in 60 μL final volume (5 × 106 parasites) (solid symbols) or at low hematocrit level (1.3%) in a final volume of 300 μL (containing 2.5 × 106 parasites) (open symbols), resulting in a 10-fold difference in hematocrit level. In both cases, the concentrations and the specific activities of the radioactive precursors were similar, ie, 10 μmol/L (3H)choline at 1.92 Ci/mmol, 2 μmol/L (3H)ethanolamine at 2.74 Ci/mmol, and trace concentration of (3H)hypoxanthine at 4 μCi/well. The results are derived from a typical experiment performed in triplicate.
Parasite growth and PL metabolism inhibition.
Lastly, we studied the correlation between the inhibition of PL metabolism (expressed as PL50) and parasite growth (IC50) for 50 compounds, which are underlined in Table 1. In this study, we excluded compounds that more likely possess other mechanisms of action, such as A2 (a widely acting sulfhydryl reagent), A9 (putrescine analog), A5 (which more likely acts as a serine analog), C1 (whose aziridin ring confers alkylating properties24), and C3, C4, and C12 (which were found to inhibit more specifically, and in a more growth-correlated way, nucleic acid metabolism than PL biosynthesis). B2 was also not considered in this study due to the absence of specific action and of correlation with growth inhibition (IC50 was more than 20-fold lower than PL50 or NA50). For this study, PL50 values obtained under high hematocrit level conditions (see above) are reported; there were not enough data obtained under low hematocrit level conditions (in which only a few compounds were investigated) to make general systematic comparisons.
As expected, the effects of these compounds on the two responses were linearly correlated (Fig 3), the slope of the regression line was 0.51, the ordinate at the origin was 1.82, and the correlation coefficient was 0.89 (which corresponds to a risk of well below 0.1%). The dotted (bisecting) line in Fig 3 shows the theoretical correlation that would be obtained if PL50equalled IC50. The molecules clearly segregated into two groups: one whose IC50 was greater than 50 μmol/L (21 molecules), appearing well distributed along the bisecting line, whereas the second group (including 29 molecules with IC50lower than 50 μmol/L) seemed to deviate from this theoretical line, with IC50 substantially lower than PL50. Among this latter group, eight compounds (see Fig 3 legend) possessing more than 12 methylene groups in their hydrophobic alkyl chain were shown to have a hematocrit-dependent effect (Table 2). Nine other compounds also probably possess the same property considering their alkyl chain lengths. Four compounds were shown to have no hematocrit-dependent effect, which was probably also the case for three others considering the methylene number present in their hydrophobic chain. This group thus mainly contains molecules (about two-thirds) with a hematocrit-dependent effect, and at equal hematocrit level, the PL50 would probably be more closely correlated with IC50. Overall, this could explain the observed deviation for this group of active compounds.

Correlation between the inhibition of PL metabolism and the inhibition of parasite growth. PL50 and IC50 (concentration producing 50% inhibition of PL synthesis and parasite growth, respectively) measured as described in the Materials and Methods were from Table 1. PL50 values correspond to high hematocrit level conditions (Table 2). The 50 compounds considered for this correlation are in bold characters and underlined in Table 1. Squares correspond to compounds for which a hematocrit-dependent effect on PL metabolism was demonstrated (▪) or probable (□). Diamonds indicate compounds for which no hematocrit-dependent effect was demonstrated (⧫) or probable (◊ ), and (×) stands for compounds of group H for which a hematocrit level effect was not determined. The dotted line corresponds to the theoretical line when PL50 = IC50 (slope = 1, ordinate at the origin = 0).
Correlation between the inhibition of PL metabolism and the inhibition of parasite growth. PL50 and IC50 (concentration producing 50% inhibition of PL synthesis and parasite growth, respectively) measured as described in the Materials and Methods were from Table 1. PL50 values correspond to high hematocrit level conditions (Table 2). The 50 compounds considered for this correlation are in bold characters and underlined in Table 1. Squares correspond to compounds for which a hematocrit-dependent effect on PL metabolism was demonstrated (▪) or probable (□). Diamonds indicate compounds for which no hematocrit-dependent effect was demonstrated (⧫) or probable (◊ ), and (×) stands for compounds of group H for which a hematocrit level effect was not determined. The dotted line corresponds to the theoretical line when PL50 = IC50 (slope = 1, ordinate at the origin = 0).
We investigated the potential reversal of inhibition of PC metabolism by a high choline concentration (200 μmol/L) to further ascertain whether these molecules act by impairing choline phospholipidic metabolism in Plasmodium-infected erythrocytes. The experiment was performed with F13, which specifically inhibited PC metabolism (PC50, 44 μmol/L). Figure 4clearly shows that a 20-fold excess of choline caused a substantial shift in the dose-response curve of PC metabolism inhibition, leading to a specific 12-fold increase in the PC50 when choline was used as radioactive precursor. By contrast, under the same conditions, no modification in ethanolamine or hypoxanthine incorporation was observed (data not shown). Similar results were obtained with G3, which led to a specific shift (>14-fold) in the PC50 towards higher concentrations (data not shown).

Effect of high choline concentration on the inhibition of PC metabolism by F13. Infected erythrocytes (5 × 106infected cells) were preincubated for 5 minutes in the presence of 10 μmol/L (•) or 200 μmol/L (○) choline. The drug was then added at the indicated concentration and after 5 minutes, and the PC biosynthesis assays were initiated by the addition of 10 μCi (3H)choline and pursued for 5 hours at 37°C.
Effect of high choline concentration on the inhibition of PC metabolism by F13. Infected erythrocytes (5 × 106infected cells) were preincubated for 5 minutes in the presence of 10 μmol/L (•) or 200 μmol/L (○) choline. The drug was then added at the indicated concentration and after 5 minutes, and the PC biosynthesis assays were initiated by the addition of 10 μCi (3H)choline and pursued for 5 hours at 37°C.
Choline transport inhibition.
We checked the effect of some compounds shown to specifically inhibit PC metabolism on choline transport into infected erythrocytes. P knowlesi-infected erythrocyte suspensions that can be obtained in great quantities and at very high parasitemia (>95%) were used for this purpose. Figure 5 shows the effects on choline transport of four alkyltrimethyl ammonium compounds (group E) in which the saturated alkyl chain had from 10 to 16 carbons. Similar action patterns were obtained for all compounds. They all competitively inhibited choline influx (under zero trans-influx measurement conditions8). The Ki values for E4 (Fig 5A), E6 (Fig 5B), E7 (Fig 5C), and E8 (Fig 5D) were 1.8, 1.1, 7.2, and 3.5 μmol/L, respectively. All of these values closely agreed with previously reported IC50 and PC50 values (Table 1). In a separate set of experiments, these compounds were tested over a wider range of concentrations. Up to 20 μmol/L, they all showed competitive behavior with similar Ki values (1.9, 0.5, 5.6, and 1.8 μmol/L, respectively), but at higher concentrations (80 and 240 μmol/L) they acted in a noncompetitive manner, with Ki values around 30 to 40 μmol/L, depending on the compound (data not shown). This distinct drug concentration-dependent behavior is probably related to the inhibition site of these alkyltrimethyl ammonium salts, depending on whether they act at the outer (competitive) or inner side (noncompetitive) of the membrane, as described for normal erythrocytes.25 26

Choline influx into P knowlesi-infected erythrocytes as a function of choline concentration in the presence of E4 (A), E6 (B), E7 (C), or E8 (D). Infected erythrocyte suspensions (98% parasitized, trophozoite stage) with 5.2 × 107cells/100 μL (0.7% final hematocrit) were preincubated for 5 minutes at 37°C in the presence of 750 μL special RPMI containing the drug (solid symbols) at 3 μmol/L (A and B) or 6 μmol/L (C and D) or no (open symbols) drug. Choline influx measurement was initiated by the rapid addition of 50 μL (3H)choline (specific activity, 0.2 Ci/mmol) at the indicated concentration. After 6 minutes of incubation at 37°C, the flux was stopped by adding 2.5 mL of ice-cold special RPMI, followed by centrifugation of a 1-mL aliquot through n-dibutyl phtalate at 4°C as described in the Materials and Methods. Nonspecific choline transport was determined in the presence of 1.2 mmol/L choline under the same conditions. The results are expressed by the double-reciprocal plot of the initial velocity of choline influx into infected erythrocytes (expressed as nanomoles per 1010 infected cells per minute) ± SEM. Each point is the mean of triplicate determinations in one typical experiment.
Choline influx into P knowlesi-infected erythrocytes as a function of choline concentration in the presence of E4 (A), E6 (B), E7 (C), or E8 (D). Infected erythrocyte suspensions (98% parasitized, trophozoite stage) with 5.2 × 107cells/100 μL (0.7% final hematocrit) were preincubated for 5 minutes at 37°C in the presence of 750 μL special RPMI containing the drug (solid symbols) at 3 μmol/L (A and B) or 6 μmol/L (C and D) or no (open symbols) drug. Choline influx measurement was initiated by the rapid addition of 50 μL (3H)choline (specific activity, 0.2 Ci/mmol) at the indicated concentration. After 6 minutes of incubation at 37°C, the flux was stopped by adding 2.5 mL of ice-cold special RPMI, followed by centrifugation of a 1-mL aliquot through n-dibutyl phtalate at 4°C as described in the Materials and Methods. Nonspecific choline transport was determined in the presence of 1.2 mmol/L choline under the same conditions. The results are expressed by the double-reciprocal plot of the initial velocity of choline influx into infected erythrocytes (expressed as nanomoles per 1010 infected cells per minute) ± SEM. Each point is the mean of triplicate determinations in one typical experiment.
Toxicity of some polar head analogs.
To investigate the specificity against other rapidly dividing eucaryotic cells, 12 compounds whose IC50 againstPlasmodium ranged from 0.8 to 110 μmol/L were tested on the viability of a lymphoblastoid cell line (SAR). After 24 hours (1 cell cycle) of contact of lymphoblastoid cells with various drug concentrations at 37°C, incorporation of (3H)thymidine into nucleic acids was monitored for 5 to 6 hours at 37°C. The results are expressed as LV50, resulting in 50% inhibition of lymphocyte viability as reflected by the inhibition of thymidine incorporation (Table 3). The LV50 was from 2.5-fold (F8) to 16,600-fold (G23) higher than the IC50, confirming the absence of any correlation between the effects on parasite growth and on lymphoblastoid cell viability.
DISCUSSION
PL are absolutely necessary for parasite membrane biogenesis,3-5 and we showed that impairment of PL metabolism was lethal to P falciparum in vitro.9-12One interest of the present systematic study was to use a large series of polar head analogs, with a wide range of in vitro antimalarial activities to determine whether antimalarial activity is mediated by PL metabolism inhibition. The results could be used to establish structure-activity relationships, potentially useful for drawing up general rules for modeling new therapeutic molecules.
Generally speaking (see for more detail our recent report13), the amine compounds in groups A, B, and C (except for N-substituted compounds with a long alkyl chain) exhibited far lower antimalarial activities (IC50 ranging from 10−3 to 10−5 mol/L) than compounds containing one (group E or F) or two (groups G and H) quaternary ammonium groups, with IC50 from 10−4 to 10−8 mol/L. Most quaternary ammonium compounds of groups E, F, and G exhibited good antimalarial activity, especially those with a long alkyl chain (containing more than 8 carbon atoms); their IC50 ranged from 2.1 μmol/L to 33 nmol/L. Compounds in group F differed from those in group E by the replacement of a methyl by a hydroxyethyl group as in choline, but their IC50 did not differ substantially. The antimalarial activities of group E, F, G, and H compounds increased as a function of the alkyl chain length, up to 10 to 12 methylene groups. Increasing the length of the alkyl chain did not further improve efficacy, at least for groups E and F. The presence of another quaternary ammonium did not significantly modify the efficiencies, except when 12 carbon atoms were present between the two quaternary ammonium groups, for which there was a fivefold decrease in IC50, whereas compounds with a short alkyl chain were found to be less efficient than the corresponding monoammonium molecules. Ten compounds belonging to group A, E, F, G, or H tested against a chloroquine resistant strain (T23) possessed the same activity, which indicates that PL metabolism inhibitors could be very useful when dealing with pharmacoresistant strains of P falciparum.
Regarding mechanism of action, compounds in group A acted specifically on PL metabolism, except for those substituted at the α position of the nitrogen with hydroxymethyl that are rather close analogs of serine. Analogs monosubstituted with methyl or ethyl groups at α or β position acted specifically on PE metabolism, whereas A7, substituted with two methyls at the α position of the nitrogen, acted more specifically on PC metabolism. This highlights the importance of the steric volume close to the N, which probably determines whether a compound acts as an ethanolamine or choline analog.
Concerning the compounds in groups E and F that specifically acted on PL metabolism, some possessed PC50 about 10-fold higher than their IC50. This concerns compounds having a long alkyl chain containing more than 10 carbon atoms, whose activity was highly dependent on the hematocrit level, ie, on the number of drug molecules per cell. At low hematocrit level, ie, under conditions similar to those used for antimalarial activity assessment, PL50 and IC50 were in close agreement (Table2). By contrast, for compounds with an alkyl chain with less than 8 to 9 carbon atoms, the hematocrit level had no effect on the PL50, regardless of the group (E, F, or G), and the IC50 and PL50 remained roughly correlated. The presence of a second quaternary ammonium (group G or H) led to high specificity, but there was a slightly weaker correlation, except for HC3.
When considering the correlation between PL metabolism impairment and parasite growth inhibition, molecules appeared to segregate into two groups (Fig 3). The first one, whose IC50 was higher than 50 μmol/L (21 molecules), appeared to be well distributed along the bisecting line (PL50 = IC50). By contrast, the second group, including 29 molecules that had a long alkyl chain and an IC50 of less than 50 μmol/L, showed a PL50that was higher than expected as compared with the IC50, likely due the hematocrit level effect related to the long hydrophobic alkyl chain (see above). For all 50 compounds, there was thus a close correlation between PL metabolism impairment and parasite growth inhibition.
Other lines of evidence that these compounds act by PL metabolism inhibition were provided by the reversal of PC metabolism inhibition by an excess of choline (Fig 4). Nevertheless, choline excess did not lead to a parallel shift in IC50 (data not shown). One possible explanation could be that these compounds could enter infected erythrocytes.
Finally, four compounds were shown to be potent inhibitors of choline transport into erythrocytes, with Ki values in the micromolar range (as compared with a Kt for choline of 8.5 μmol/L8). The Ki values were in very close agreement with the PC50, indicating that PC50provides an adequate index of the inhibitory effect on the choline transporter.
Overall, these results (good IC50/PC50/Ki of choline transport correlation, reversal of de novo PC biosynthesis by choline excess) show that choline entry and incorporation into PC is probably the target of these analogs in infected erythrocytes. In particular, the effect on the second enzymatic step of the de novo PC biosynthesis (choline kinase) was ruled out, because substantial inhibition of choline kinase by E4, G3, or HC3 only occurred at millimolar concentration, ie, three orders of magnitude higher than their IC50 or PC50.11 27
In P falciparum-infected erythrocytes, choline is incorporated via a specific carrier showing high affinity for choline (Kt = 8.5 μmol/L) and whose characteristics are close to those of normal erythrocytes, except for a 10-fold increase in Vm.8 A probable absence of stereoselectivity of this carrier was suggested using α or β methyl choline stereoisomers.28 On the other hand, there is an induced permeability pathway with very broad specificity to various unrelated solutes, preferably anions, but also neutral or to a less extent, cationic solutes, because this pathway could incorporate choline at very high concentrations (millimolar).29 This latter pathway has been described as a stereoselective channel protein containing a hydrophobic region and a positive charge or dipole to provide anion selectivity.30 The pharmacologic effect observed in our study, notably with quaternary ammonium salts, concerned inhibition of high-affinity choline carrier of infected erythrocytes, considering, for instance, the imperative cationic charge requirement for antimalarial activity and PC metabolism inhibition (see the total inefficiency of D5 in which the quaternary ammonium was replaced by the tetrasubstituted carbon) or also specificity of inhibition of PC metabolism versus PE, nucleic acid (Table 1), or protein synthesis (Ancelin et al10 and data not shown). This latter characteristic as well as the permanent charge of quaternary ammonium also rule out any lysosomotropic effect previously reported for some N-dodecyl substituted tertiary amines that impaired parasite protein synthesis.31
As for the hematocrit level effect, there is also a pivotal point for the choline transport inhibition pattern as a function of chain length, because the lowest Ki values for choline transport inhibition correspond to 10 to 12 carbons (Fig 5). The carbon chains of these compounds probably form hydrophobic associations with the transporter so that the alkyl group extends from the trimethyl ammonium moiety over a length of approximately 12 carbon atoms. The increase in the Ki values at 14 carbon chain length suggests that the end of the hydrophobic alkyl group may butt against a hydrophilic domain and be repelled by it.
Potent antimalarials are therefore substances having a small quaternary ammonium group with a long hydrophobic chain, which could likely combine with an anionic site and a long hydrophobic region (corresponding to 10 to 12 methylenes) of the choline transporter. Substances with two quaternary ammonium groups are as potent as (up to 10 methylenes) or slightly more potent (12 methylenes) than the corresponding monoquaternary compounds.
Nevertheless, a crucial problem in drug development is the specificity required to achieve an acceptable therapeutic index. The usually much higher toxic effect on the malarial parasite than on the lymphoblastoid cell line, as well as the total absence of correlation between IC50 and LV50 (Table 3), more likely indicates that the structural requirements for inhibition of PL metabolism are highly specific to infected cells and/or that the malarial parasite is more dependent on this metabolism.
A second potential problem with choline analogs concerns involvement of choline as precursor of the neurotransmitter, acetylcholine. Some of the present data indicate that the structural requirements to inhibit choline entry into infected erythrocytes differ from that to inhibit high-affinity choline transport (HACT) in synaptosomes.32 33
First, the cholinergic compound hemicholinium (HC3) showed a far lower IC50 in synaptosomes (0.02 μmol/L) than the bisquaternary compound G3 (1.5 μmol/L).33 By contrast, against P falciparum, the activities of both of them are very close (4 μmol/L and 1.7 μmol/L, respectively). Because the distance between the two nitrogen atoms in HC3 is very close to that spanned by the 10 methylene groups of G3, the presence of aromatic rings between the two quaternary ammonium groups and/or the possibility of hydrogen-bonding through the HC3 hydroxyl group to the binding site do not seem to be involved in its antimalarial effect on P falciparum, contrary to its cholinergic effect on synaptosomes.32
Secondly, substantial differences between the choline carrier in erythrocytes and synaptosome HACT (see Fisher and Hanin34for review) also concern the steric fit of analogs at the active site. For small choline analogs, slightly increasing the steric hindrance of the polar head (N substitutions) is dramatic for the erythrocyte facilitated choline transport system but not for the HACT system.35 N-ethylcholine (F1) is bound almost as well as choline by the HACT, but 100-fold more weakly than choline through erythrocyte choline transport,35 and it was also weakly active against P falciparum (IC50, >300 μmol/L). Finally, using methyl choline stereoisomers, we have also shown other characteristics distinct from that of synaptosomes,36 such as the absence of stereospecificity and the opposite effect of methyl substitution at the α or β position of the nitrogen.28 The structural requirements for inhibition of choline incorporation into P falciparum-infected erythrocytes are thus very specific and differ from the requirements for inhibition of choline uptake into synaptosomes and also into many other animal cells and tissues (see Lerner37 for review).
Hence, the antimalarial activity of the 77 polar head analogs is mediated by inhibition of PL biosynthesis. PL metabolism of P falciparum-infected erythrocytes, especially de novo PC biosynthesis from choline, is thus a quite realistic target for a new malaria chemotherapy, even in cases of polypharmacoresistance. The highest antimalarial activities measured here were around 0.1 μmol/L (E10, E20, F8, G4, and G23) and as low as 33 nmol/L for E13. Moreover, the structure/activity relationships already highlighted13provide general rules for improving this activity and facilitate modeling of new therapeutic molecules.



ACKNOWLEDGMENT
The authors owe special thanks to Prof J.W. Kosh (University of South Carolina, Columbia, SC) for providing selenium choline and to F. Vialettes for his skilled technical assistance.
A compound was defined as PL-specific when it affected the metabolism of only one PL, either PC or PE from choline or ethanolamine, respectively, without any simultaneous effect on the metabolism of the other PL, or on nucleic acid synthesis (ie, when the PL50 was at least 2.5-fold lower than the NA50).
A compound was considered to show a good correlation between PL metabolism and parasite growth inhibition when its PL50 was in close agreement with its IC 50 , ie, when there was less than 2.5-fold difference between values.
Supported by the UNDP/World Bank/WHO special program for Research and Training in Tropical Diseases (Grant No. 950165), the Commission of the European Communities (INCO-DC, PL-950529:IC18-CT960056), the CNRS (GDR No.1077, Etude des parasites pathogènes), the Ministère de l'Enseignement Supérieur et de la Recherche (DPST No. 5 and MENESR-DGA/DSP and AUPELF-UREF ARC no. X/7.10.04/Palu 95), and the VIRBAC Laboratories.
Address reprint requests to Marie L. Ancelin, PhD, CNRS UMR 5539, Department of Biologie-Santé, CP 107, UM II, Place E. Bataillon, 34095 Montpellier Cedex 5, France.
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. section 1734 solely to indicate this fact.