Abstract
Platelets, the small, anucleate blood cells that originate from megakaryocytes in the bone marrow, are typically associated with coagulation. However, it is now apparent that platelets are more multifaceted than originally thought, with their function extending beyond their traditional role in hemostasis to acting as important mediators of brain function. In this review, we outline the broad repertoire of platelet function in the central nervous system, focusing on the similarities between platelets and neurons. We also summarize the role that platelets play in the pathophysiology of various neurological diseases, with a particular focus on neuroinflammation and neurodegeneration. Finally, we highlight the exciting prospect of harnessing the unique features of the platelet proteome and extracellular vesicles, which are rich in neurotrophic, antioxidative, and antiinflammatory factors, for the development of novel neuroprotective and neuroregenerative interventions to treat various neurodegenerative and traumatic pathologies.
Functions of platelets: beyond hemostasis
Platelets, the smallest blood cells, are derived from megakaryocytes and are most well known for their role in clot formation. However, less than one-tenth of the circulating platelets are required to facilitate hemostasis, suggesting that these cells likely fulfill additional functions.1-4 Platelets are now recognized as essential components of the immune system, capable of participating in both innate and adaptive immune responses.1-4 They contribute to innate immunity through their ability to release a plethora of inflammatory molecules upon activation, which attract or modulate effector cells and direct leukocytes toward sites of inflammation. Platelets also express Toll-like receptors, which can directly bind pathogens via the detection of pathogen-associated molecule patterns.5 In addition, growing evidence suggests that they mediate adaptive immune responses via their expression of functional CD40L.6 Platelets can also deliver antigens to antigen-presenting cells,7 process and present antigens themselves,8 and influence T-cell responses.9
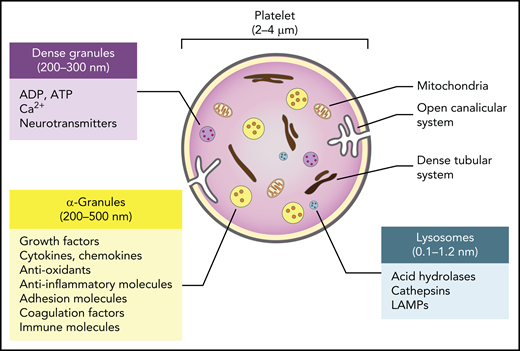
Despite containing no nucleus, and therefore playing no role in transcription, platelets contain messenger RNA (mRNA), a functional proteasome containing antigen-presentation machinery,10 and a surprisingly complete set of organelles and storage granules, including mitochondria, lysosomes, α (α)- and dense granules rich in small molecules and trophic factors, a dense tubular system analogous to the endoplasmic reticulum, and a highly invaginated plasma membrane system known as the open canalicular system (Figure 1). The human platelet proteome is currently estimated to contain approximately 5200 proteins.11 Although a recent study revealed a high similarity between the transcriptomes of platelets and megakaryocytes, many transcripts are not expressed in platelets, suggesting a redistribution of RNA species inherited from megakaryocytes following platelet shedding in the blood.11 There is, however, a good relationship between the platelet transcriptome and proteome, indicating the capacity for platelets to carry out translational processes.11 In addition, the open canalicular system, which makes up approximately 1% of the platelet volume, is filled with proteins captured from the plasma by diffusion and endocytosis.
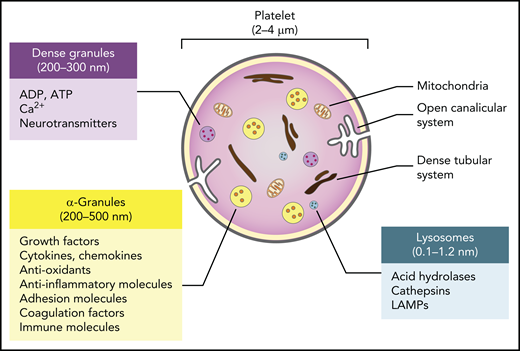
Major features and biological cargo of platelets. Platelets contain various organelles and storage granules, including mitochondria, lysosomes, α- and dense granules, a dense tubular system, and a highly invaginated plasma membrane system known as the open canalicular system. The α- and dense granules and lysosomes contain a plethora of small molecules and trophic factors that are released upon activation.
Major features and biological cargo of platelets. Platelets contain various organelles and storage granules, including mitochondria, lysosomes, α- and dense granules, a dense tubular system, and a highly invaginated plasma membrane system known as the open canalicular system. The α- and dense granules and lysosomes contain a plethora of small molecules and trophic factors that are released upon activation.
The largest and most abundant of the platelet granules are the α-granules. Between 200 and 500 nm in diameter and numbering approximately 50 to 80 per cell, these granules resemble the large dense-core vesicles of neurons and contain proteins such as growth factors, cytokines, antioxidants, and adhesion molecules that are either synthesized in the megakaryocytes before platelet segmentation or taken up from the plasma by exocytosis. The dense granules are smaller (200-300 nm in diameter), approximately 10-fold fewer in number than α-granules, and resemble the small dense-core vesicles of neurons. These granules primarily contain small molecules such as serotonin and other neurotransmitters, adenosine triphosphate, adenosine diphosphate (ADP), and calcium, which are taken up from extracellular pools. Platelets also typically have 1 to 2 lysosomes per cell that contain acid hydrolases, cathepsins, and lysosomal-associated membrane glycoproteins.
To fulfill particular functions, platelets must first be activated. Distinct subsets of proteins are secreted in response to specific activation stimuli, as evidenced by their characteristic response to 3 common agonists: ADP, collagen, and thrombin receptor-activating peptide.12 In addition, the existence of subpopulations of α-granules, in which unique subsets of proteins are stored and released by stimulation with specific agonists, further highlights the finely tuned mechanism by which bioactive molecules are released by platelets in a context-dependent manner.13 In addition to the direct secretion of bioactive molecules from granules, platelets also release extracellular vesicles (EVs).14 First described as “platelet dust” in the 1960s,15 EVs are emerging as important mediators of intercellular communication. The majority of EVs in the blood are platelet- or megakaryocyte-derived.16,17 Although resting platelets constitutively produce EVs,18 activation increases their number and defines their specific cargo.19 Platelets are believed to predominantly release 2 types of EVs: platelet microvesicles (PMVs) and exosomes.14 PMVs (0.1-1 µm in diameter) are shed from the plasma membrane20 and, like platelets, carry selective subsets of RNA, proteins, and organelles such as mitochondria, with their heterogeneity being dictated by the activation stimulus which triggers their formation.21,22 However, the precise mechanism by which they selectively package and release their biological cargo remains unclear. Exosomes (30-100 nm in diameter) are smaller than PMVs and are derived from endosomal multivesicular bodies and α-granules.23 Interestingly, activated platelets also release respiratory-competent mitochondria as free organelles that can act as damage-associated molecular patterns in several pathologies.24
In addition to their role in the periphery, it is becoming increasingly clear that platelets also contribute to both the degeneration and regeneration of the central nervous system (CNS). Both platelets and the EVs they release are small enough to traverse the microcapillaries spanning the brain under both healthy and pathological conditions. This enables crosstalk with neural cells via a number of mechanisms, either indirectly via the secretion of bioactive molecules from platelet granules, exosomes, or PMVs, or directly by local receptor-mediated interactions.25 Interestingly, platelets also exhibit a remarkable number of similarities to neurons, further highlighting the potential for crosstalk between these cells and the brain.
Similarities between platelets and neurons may facilitate their crosstalk
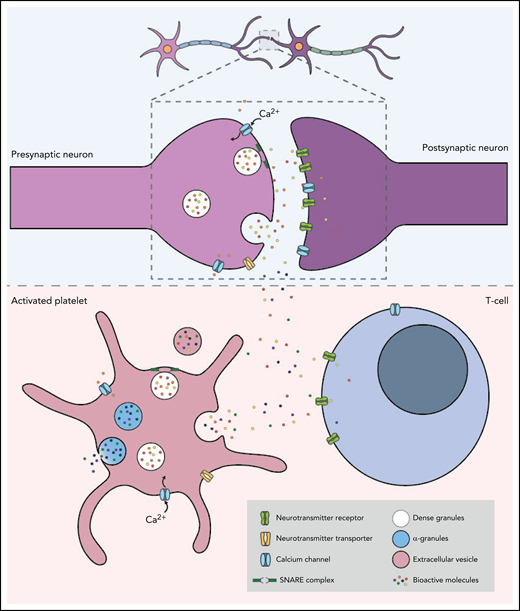
Despite their different embryonic origins, with neurons arising from the ectoderm and platelets from megakaryocytes that originate from the mesoderm, it has been proposed that, in many ways, the communication between platelets and other immune cells, including neutrophils, monocytes, and clusters of differentiation (CD)8+ T cells, mirrors the interaction between pre- and postsynaptic neuronal terminals26-28 (Figure 2). These similarities may facilitate the crosstalk between platelets and neurons in the wide range of neurodegenerative disorders in which platelet dysfunction has been implicated. In a mechanism analogous to that which occurs at the presynapse of neurons, calcium-dependent activation of the platelet secretory machinery triggers the fusion of internal vesicles with the plasma membrane via the specific, highly conserved docking molecules, SNAREs (soluble NSF attachment protein receptors), resulting in the rapid vesicular release of neurotransmitters.29 There is, however, a marked difference in the kinetics of signal transduction and exocytosis between platelets and neurons, with neurotransmitter release occurring 10 000 times faster in neurons.29,30 Although increases in intracellular Ca2+ trigger exocytosis in both cell types, the magnitude and mechanism of the calcium signal generation differ significantly. In neurons, membrane depolarization results in Ca2+ influx via Ca2+ channels, which leads to a local Ca2+ increase of ∼200 mmol/L and triggers the exocytosis of nearby vesicles. In contrast, platelet granule secretion is mediated by the activation of extracellular receptors, which increases the concentration of intracellular cytosolic Ca2+ (to ∼10 mmol/L) and activates protein kinase C. Moreover, unlike platelets which typically undergo irreversible aggregation following activation, neurons rapidly return to their resting state and can be reactivated following a refractory period of approximately 1 ms.
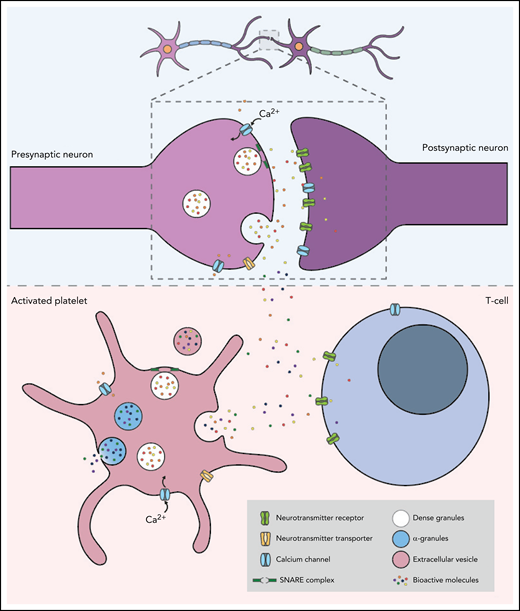
Platelets and neurons share many similarities. The top panel shows the communication between a presynaptic and a postsynaptic neuron. Neurons are activated by calcium signaling, which triggers the release of neurotransmitters such as serotonin, dopamine, epinephrine, histamine, glutamate, and GABA. The neurotransmitters then bind to the corresponding receptors on the postsynaptic neuron to complete neurotransmission. The lower panel shows an activated platelet with dense granules containing neurotransmitters, adenosine triphosphate, adenosine diphosphate, and calcium, and α-granules containing growth factors, cytokines, antioxidants, and adhesion molecules. Similar to the mechanism which occurs at the presynapse of neurons, calcium-dependent activation of platelets triggers the fusion of the granules with the plasma membrane via highly conserved docking molecules called SNAREs. This results in the rapid release of neurotransmitters, which bind to corresponding receptors expressed on the surface of T cells.
Platelets and neurons share many similarities. The top panel shows the communication between a presynaptic and a postsynaptic neuron. Neurons are activated by calcium signaling, which triggers the release of neurotransmitters such as serotonin, dopamine, epinephrine, histamine, glutamate, and GABA. The neurotransmitters then bind to the corresponding receptors on the postsynaptic neuron to complete neurotransmission. The lower panel shows an activated platelet with dense granules containing neurotransmitters, adenosine triphosphate, adenosine diphosphate, and calcium, and α-granules containing growth factors, cytokines, antioxidants, and adhesion molecules. Similar to the mechanism which occurs at the presynapse of neurons, calcium-dependent activation of platelets triggers the fusion of the granules with the plasma membrane via highly conserved docking molecules called SNAREs. This results in the rapid release of neurotransmitters, which bind to corresponding receptors expressed on the surface of T cells.
Platelets also carry a plethora of neurotransmitters crucial for intercellular communication between neurons, together with the receptors and transporters of these molecules, further highlighting the crosstalk and homology between the periphery and the CNS. The most abundant of these is the monoamine serotonin.31 Platelets take up serotonin from the plasma via the serotonin transporter and store it in dense granules. The release of serotonin from dense granules is a marker of the degree of platelet activation while also contributing to platelet aggregation, the recruitment of circulating platelets, and the promotion of coagulation. In addition to serotonin, platelets carry several other neurotransmitters crucial for the intercellular communication between neurons, including other biogenic amines such as epinephrine, dopamine, and histamine, the excitatory neurotransmitter glutamate, and the inhibitory neurotransmitter γ-aminobutyric acid (GABA). Glutamate is the most abundant excitatory neurotransmitter in the brain, and as in neurons, substrate-induced uptake of glutamate in human platelets most likely occurs via glutamate receptors.32 Platelets store significant amounts of GABA, although the concentrations are approximately 30% lower than in neurons.33
Platelets also transport unusually high concentrations of several other proteins with important neuronal functions. The protein reelin, which regulates cell migration and synaptic plasticity, is carried by platelets, where it modifies platelet cytoskeletal dynamics and contributes to arterial thrombosis.34,35 Following platelet activation, reelin is secreted and binds to the amyloid precursor protein (APP) and apolipoprotein E receptor 2 to induce signaling.35 Interestingly, brain-derived neurotrophic factor (BDNF), a secretory protein that is primarily known for its role in regulating the development and function of neural circuits, accumulates in human platelets at 100- to 1000-fold higher levels than in neurons.36 During hemostasis, BDNF is released by platelets and modulates thrombosis.37,38 Surprisingly, however, BDNF is absent in mouse platelets and serum,39 suggesting that its biological role in platelets may be compensated by other platelet-derived growth factors. Other well-characterized platelet trophic factors that are important in mediating brain function include growth factors (eg, platelet-derived growth factor-AA and -BB, transforming growth factor-β, epidermal growth factor, vascular endothelial growth factor, and basic fibroblast growth factor), chemokines (eg, CXCL4 [platelet factor 4], CCL5 [chemokine ligand 5], and CCL3), and various antioxidants such as glutathione peroxidase.40-42 However, despite the similarities between neurons and platelets, why these cells have evolved to resemble each other remains unclear.
Platelets mediate neural plasticity
Neural plasticity refers to the ability of neural networks within the brain to change through growth or reorganization in response to experience. One way in which the brain retains lifelong plasticity is via the continuous addition of newly generated neurons into the existing circuitry, a process known as adult neurogenesis. The neurogenic niches are highly vascularized, with the neural stem cells lying in direct contact with blood vessels.43-45 It has even been suggested that platelets should be considered as part of the neurovascular unit,46 which typically includes neurons, glial cells, neural precursor cells (NPCs), endothelial cells, vascular smooth muscle cells, and pericytes.
Several studies have shown that platelets can modulate NPC proliferation and neurogenesis in the subventricular zone (SVZ), although this has only been studied in vitro or in the context of brain injury. Hayon and colleagues were the first to demonstrate that platelet microparticles can increase embryonic NPC proliferation, survival, and neuronal-lineage differentiation potential in vitro.47 Another study found that platelet lysate increased the number of in vitro proliferating adult rat SVZ-derived NPCs by enhancing their survival via a reduction in apoptosis without affecting their proliferative or lineage-differentiation capacity.46 Neurogenesis-enhancing effects of platelets have also been observed in animal models of stroke.46,48,49 Recently, platelets were found to regulate in vitro hippocampal neurogenesis, with activated platelets increasing the proliferation and neuronal lineage differentiation potential of the primary dentate gyrus-derived NPCs.50 Physical exercise is one of the strongest physiological mediators of hippocampal neurogenesis. Platelets were found to contribute to a running-induced increase in mouse hippocampal neurogenesis, with induction of thrombocytopenia abolishing the exercise-induced increase in NPC proliferation, indicating that platelets are required for this response.50 However, no difference in baseline neurogenesis amounts was observed between the sedentary platelet-depleted and control mice. Interestingly, a proteomic comparison revealed that none of the classical platelet activation markers, such as the activation-specific glycoprotein (GP) IIb and IIIa receptor,51 was differentially expressed following exercise, suggesting that exercise-induced platelet activation is a distinct process.50
Another way in which the brain retains plasticity is via changes that occur at synapses, the junctions that allow for communication between neurons. Neurovascular damage, such as traumatic brain injury (TBI), and neurovascular diseases, such as stroke, typically result in neuroinflammation accompanied by an increase in neuronal plasticity. Dukhinova and colleagues52 proposed that dynamic platelet–neuron synapse-like structures form after brain injury, which induces platelets to release serotonin,53 thereby stimulating neurite growth and the formation of new synapses.52 Platelets were also found to upregulate the expression of several genes involved in neuronal synaptic plasticity, stimulating the formation of dendritic spines and new synapses.52,54 In support of these findings, an earlier study found that platelet-activating factor can induce hippocampal long-term potentiation, a form of activity-driven synaptic plasticity that involves a persistent strengthening of synapses, leading to a long-lasting increase in signal transmission between neurons.55
Platelets and neuroinflammation
In the previous sections, we discussed the physiological links between neurons and platelets. In the second half of this review, we will shift our focus to disease processes. Neuroinflammation, which contributes to the pathophysiology of various neurological diseases, comprises a cascade of accumulating damage encompassing cellular and molecular components, including the generation of reactive oxygen species, the release of cytokines and chemokines, microglial and astrocytic activation, neuronal protein unfolding and aggregation, and neuronal cell death.56,57 It is now becoming clear that systemic inflammation, neurovascular dysfunction, and coagulopathy coexist in neurological pathologies, including Alzheimer’s disease (AD), Parkinson’s disease (PD), and multiple sclerosis (MS),58,59 highlighting a likely role of the blood compartment in the detrimental inflammatory aspect of neurological diseases. The concept that the blood system is involved in the pathophysiology of brain neuroinflammation is supported by the high level of vascularization of the CNS parenchyma, which facilitates crosstalk between the blood and the brain, and the loss of integrity of the blood–brain barrier (BBB) that occurs in various chronic neurodegenerative disorders and as a consequence of brain trauma.60 Among the multiple cellular and protein components of blood, platelets are thought to provide a link between the blood and the brain in disease,25,61 as evidenced by platelet invasion and deposition in brain lesions.59
Platelets play a critical role in inflammatory processes and immune responses62 but also exhibit antiinflammatory potential through the regulation of macrophage function and regulatory T cells and the secretion of antiinflammatory factors.63 The functional capacity of platelets to act as inflammatory mediators (and hence to potentially modulate neuroinflammation) is supported by the fact that they are the first cells to reach sites of injury or inflammation. Platelets are therefore highly reactive to discrete changes that affect the plasma coagulation and complement systems, leading to their activation, exposure of functional membrane receptors,64 degranulation, and the release of trophic factors, cytokines, and chemokines.64 Under inflammatory states, platelets also release EVs that act as complementary proinflammatory mediators, capable of infiltrating tissues65 and capturing and activating neutrophils.66
Various components expressed by activated platelets and platelet EVs can promote leukocyte–endothelial interactions and trigger inflammatory processes.66 Those components include phosphatidylserine, a coagulant phospholipid, a range of surface glycoproteins, proinflammatory cytokines and chemokines, as well as matrix metalloproteinases.59,67 In line with this, a recent study found that the activated platelet-mediated release of pathogenic serotonin after exposure to systemic immune complexes is a critical mediator of shock.68
The interaction between platelets and the CNS involves crosstalk between activated platelets and vascular endothelial cell receptors and the recruitment of leukocytes and neutrophils from the blood circulation. Strikingly, activated platelets have been shown to affect the BBB integrity and infiltrate the brain69 in conditions such as stroke70 and MS,71 and interact with the cellular components of the BBB matrix (endothelial cells, pericytes, perivascular microglia, and astrocytes).72 Soluble CD40L, for example, was found to be a key contributor to astrocytic and microglial activation in hypertension, which increases the expression of adhesion molecules on the brain endothelium and results in deposition of platelets in the brain, leading to neuroinflammation and neuronal injury.73 Platelet–neuron associations have also been observed in the hippocampal parenchyma in animal models of MS, an observation with potential therapeutic relevance for treating the neuropsychiatric symptoms of the disease.71 Moreover, direct contact with brain glycolipid structures present in the membranes of astrocytes further activates platelets and stimulates neuroinflammation,53 leading to immune cell recruitment into the brain parenchyma74 and the simultaneous release of trophic factors.27,53,72 Interestingly, this cascade of events creates a microenvironment where proinflammatory cytokines, neurotrophic factors, and neurotransmitters coexist, which, in response to CNS damage, can stimulate new synapse formation and axonal regrowth.27 A specific role in the modulation of neuroinflammation has been assigned to the α-granule chemoattractants CXCL4 and CCL5 through monocyte recruitment, or MIP-1a (macrophage inflammatory protein-1a)67,75 because of interaction with immune cells, BBB cellular components, astrocytes and microglia, as well as neurons themselves. Together, these observations highlight the contribution of various platelet factors in neuroinflammation and have provided incentives for preclinical studies where the protective effect of antiplatelet adjunct therapy, such as aspirin, has been examined.76 The potential of various new classes of oral antiplatelet agents is being evaluated in preclinical and clinical studies, most notably to diminish ischemic stroke recurrence, albeit the risk of increasing intracranial bleeding needs to be carefully considered. These agents include vorapaxar, which blocks PAR-1 (protease-activated receptor-1); clopidogrel and ticagrelor, which inhibit P2Y12; revacept, a GPVI (glycoprotein VI) fusion protein; ACT101, a monoclonal anti-GPVI antibody; caplacizumab, an anti-von Willebrand factor nanobody; and cilostazol, a PDE-3 (phosphodiesterase-3) inhibitor of platelet aggregation. Another antiplatelet agent, ibudilast, which inhibits PDE-4, has been shown to slow brain atrophy in MS patients, consistent with the increased detrimental prothrombotic effect of platelets. Further studies are, however, required to delineate the therapeutic role of antiplatelet therapies in neurological disorders.75
Platelet dysfunction in neurodegenerative diseases
It is clear that neurodegenerative diseases not only involve the cells within the CNS, but that systemic mediators such as platelets also play an important role in their manifestation, with platelet dysfunction being observed in disorders including AD, PD, Huntington’s disease (HD), amyotrophic lateral sclerosis (ALS), and MS. As such, platelets have been described as “circulating mirrors of neurons,”77 providing an easily accessible peripheral biomarker to monitor the onset and progression of these conditions. The key alterations in the physical parameters and molecular pathways of platelets in these conditions are summarized in Table 1.
The links between platelet dysfunction and neurodegeneration have been reviewed in detail elsewhere.61,77-80 Therefore, in the following section, we have chosen to highlight the links between platelets and AD, an age-related neurodegenerative disorder and the most prevalent form of dementia. The pathological hallmarks of this disease are extracellular deposits of amyloid-β (Aβ), intraneuronal aggregates of hyperphosphorylated τ, activated microglia, reactive astrocytes, dystrophic neurites, inflammation, and oxidative stress. Several studies have linked both normal platelet function and platelet dysfunction to the development of AD. Platelets are the primary source of circulating APP from which they can produce and subsequently secrete Aβ.81,82 Outside the CNS, platelets are the main source of Aβ.83,84 Both Aβ and all 3 isoforms of APP are expressed in megakaryocytes and stored in platelet α-granules from which they are released upon activation. Whereas neurons produce significant amounts of the Aβ42 peptide, platelets mainly produce Aβ40, which they release upon activation, contributing to approximately 90% of the Aβ in the blood.82,85 Unlike neurons, which predominantly process APP via the β-secretase pathway, platelets primarily process APP and release Aβ through α-secretase via the regulated secretory vesicle pathway.86,87 Nonetheless, platelets contain all 3 secretases and, in principle, could therefore generate all APP cleavage products.88 The physiological role of APP and its cleavage products in platelets remains unclear, although the antimicrobial effect of Aβ is clearly established, suggesting that it may act as a normal component of the innate immune system.89,90 Circulating Aβ can also cross the BBB.91 It was recently shown that platelets accelerate the amyloidosis process by disrupting the permeability of the BBB and accelerating the accumulation of Aβ in the brain parenchyma, thereby leading to the manifestation of AD in animal models.92-94 Together, a number of studies indicate that, in AD, platelets become hyperactive and release and interact with Aβ to accelerate disease progression,92,95,96 suggesting that the disease may, at least in part, be a slowly developing thrombohemorrhagic disorder.97
Parabiosis between transgenic AD and wild-type mice has been shown to increase Aβ in the vessel walls and brain parenchyma and drive the formation of AD-like protein aggregates in the brain of the latter animals.98 Similarly, another study found that plasma from young mice reduced both Aβ and τ pathologies and neuroinflammation and ameliorated cognitive impairment in transgenic AD mice.99 In contrast, a study using human APP transgenic mice found that exposure to young blood did not alter amyloid plaque deposition.100 Although clinical trials in which AD patients receive young plasma are currently underway,101,102 the contribution of platelets to the properties and composition of this young plasma remains unknown.
In addition to the role of platelet-derived Aβ in AD pathogenesis, platelets may also serve as a peripheral biomarker. The ratio of platelet APP isoforms is significantly altered in AD patients, enabling the detection of conversion from mild cognitive impairment to AD.103 Although less well-studied, τ is also present in platelets. The ratio of high molecular weight forms to low molecular weight monomeric τ was found to be significantly higher in the platelets of AD patients.104,105 More recently, the levels of platelet Ser396/404 phosphorylated τ and Thr231 phosphorylated τ were found to be significantly higher in mild cognitive impairment patients and could be used to distinguish between them and normal subjects.106
Despite considerable research, it remains unclear whether platelet dysfunction occurs before or because of the neurodegenerative process. Nevertheless, dampening platelet activation may represent a therapeutic approach to AD. Several studies have investigated antiplatelet agents as potential therapeutic compounds in AD. Of these, the most well-studied is low-dose aspirin, which has been shown to suppress the secretion of APP from human platelets.107 Unfortunately, however, the majority of studies have indicated that it is not effective in preventing or treating the disease.108,109 In contrast, the effects of another antiplatelet agent, cilostazol, appear to be more positive.110-112 Further studies are therefore required to fully elucidate the role of platelet activation in AD pathophysiology in order to develop new diagnostic and prognostic biomarkers as well as therapeutic approaches for the disease.
In addition to the neurodegenerative diseases discussed above, platelet dysfunction has been implicated in the neurological manifestations of other conditions. Immune thrombocytopenia is associated with neurological complications following many viral infections, including hepatitis C virus, human immunodeficiency virus, cytomegalovirus, Epstein-Barr virus, hantavirus, herpes virus, severe acute respiratory syndrome coronavirus, dengue virus, and Zika virus, potentially via platelet-induced BBB breakdown.113 Likewise, platelets have been shown to contribute to BBB breakdown in cerebral malaria, resulting in severe neurological complications.114 It has also been proposed that the increased platelet activation observed in diabetic patients contributes to an increased risk of developing AD via the release of inflammatory factors and Aβ.115 Moreover, essential thrombocythemia, an acquired myeloproliferative neoplasm that leads to elevated platelet concentrations, is associated with usually transient neurological symptoms, including blurred vision, headache, tinnitus, and dizziness. Although antiplatelet therapy is often used to treat these conditions, further research is required to fully elucidate the effectiveness of this approach.
Looking to the future: harnessing platelet derivatives as potential therapeutics
Healthy platelet granules contain an amazing reservoir of potent antiinflammatory, neuroprotective, and antioxidative molecules63 that can potentially be harnessed for the treatment of neurodegenerative conditions. The translational rationale for using platelets in this way is linked not only to their high content of potent biomolecules but also to the fact that clinical platelet concentrates are readily available in many countries, thereby facilitating access to this potential biotherapy.116 In addition, the efficacy of platelet concentrates that are pathogen-reduced but no longer suitable for direct transfusion for cell therapy and regenerative medicine has been demonstrated in cell culture and animal models.116,117 Processing of platelets as source material of human platelet lysate (HPL) biologicals also benefits from the technical and regulatory experience gained with other blood-derived medicinal products.118
Recent preclinical work has evaluated human platelet-rich plasma or platelet concentrates as source materials for neurotrophin-rich platelet lysates and EV preparations for the treatment of CNS diseases40,49,117,119-125 (Table 2). For example, the administration of HPL to the lateral ventricles of rats subjected to focal ischemia induced by permanent distal middle cerebral artery occlusion significantly enhanced the proliferation of endogenous neural stem cells and angiogenesis in the SVZ and in the perilesion cortex and improved neurobehavioral outcomes.49 Similarly, in an AD model, intranasal administration of HPL exerted a neuroprotective action, which led to decreased Aβ deposition, τ phosphorylation, and astrocyte reactivity, thereby preventing synaptic loss and stimulating global improvements in anxiety, learning, and memory behaviors.120 Intranasal delivery of HPL also decreased neuroinflammation and improved motor performance in a mouse model of PD.122 Consistent with these data, the intranasal administration of purified HPL resulted in robust protection of the dopaminergic neurons in the substantia nigra and striatum.119 This platelet lysate diffused to various brain areas, including the olfactory bulb, striatum, and cortex, without detectable induction of neuroinflammation, suggesting that its intranasal delivery may offer therapeutic potential against several neurodegenerative diseases.119 The multifaceted therapeutic potential of HPL has also recently been confirmed in models of TBI.40 The HPL was first applied topically in the lesioned area to mimic a severe TBI in which there would be brain access, followed by daily intranasal administration for 6 days after injury. This treatment significantly improved motor function, downregulated proinflammatory genes, controlled the level of reactive oxygen species, and reduced the loss of cortical synaptic proteins.
The use of platelet lysates or purified fractions to treat ALS has also been investigated.124 Platelet lysate treatment of NSC-34 (a spinal cord and neuroblastoma hybrid cell line) motor neurons exposed to staurosporine and menadione toxins resulted in strong neuroprotection.123 A HPL and its fractions separated based on molecular mass also induced protein kinase B (also known as Akt)-dependent neuroprotection as well as a strong antiapoptotic and antiferroptotic action in neuronal cell cultures.126 The low molecular weight (<3 kDa) fraction had glutathione peroxidase 4-dependent antiferroptotic properties, suggesting a protective role of platelet factors other than the higher molecular weight neurotrophins. Consistent with the in vitro findings, the lifespan of superoxide dismutase 1G86R mice, a model of ALS, was significantly increased by intracerebroventricular delivery of the complete lysate and intranasal administration of the <3 kDa fraction,126 expanding the scope of potential therapeutic applications.
There is also increasing interest in therapeutic applications of platelet EVs as drug delivery systems due to their ability to transport trophic factors, cross tissue barriers, diffuse into tissues, and target cells. The possibility of using EVs, rather than free molecules or nanocarriers,127 as neurotrophic factor-rich therapeutic formulations against CNS disorders has been evidenced using mesenchymal stem cell EVs in preclinical models of neurological diseases.128 Clinical-grade platelet concentrates and HPL contain as many as 1012 EVs/mL,129,130 with a mean diameter of 50 to 300 nm and a slightly negative ζ potential that can be exploited for brain diffusion and the delivery of neurotrophic factors.130 Similar to platelets, EVs carry growth factors, cytokines, and chemokines, as well as antiinflammatory molecules and microRNAs,131 although whether they play a critical role in the neuroprotective function of HPL remains unclear, as does their impact on HPL safety for brain administration.131,132 However, compared with unfractionated HPLs, EV administration may decrease protein overload in the CNS, prolong protection and the release of trophic factors, and support the targeting of pathological brain tissues.133 Preliminary evidence has revealed that EVs purified from various HPLs are nontoxic and capable of stimulating neuroblastoma cell healing as well as favoring network formation in primary cortical neuronal cultures.130 It has also been found that platelet EVs promote the in vitro proliferation and differentiation of neural stem cells47 and functionally stimulate neurogenesis when topically applied with a biodegradable polymer in an in vivo stroke model, further demonstrating the ability of EVs to diffuse within the brain.48 Together, these data support the need for further evaluation of platelet EVs as a standalone biotherapy or drug delivery vehicle in neuroregenerative medicine.
Future directions
As summarized above, there is now considerable evidence that the functions of platelets extend well beyond their canonical role in hemostasis. Platelet dysfunction has been implicated in several neurodegenerative and neuroinflammatory conditions, opening fascinating new perspectives for a deeper understanding of the pathophysiology of these disorders. However, whether platelet dysfunction initiates the onset of pathophysiology or arises as a consequence of it remains unclear. Nevertheless, knowledge of the crosstalk between platelets and the brain parenchyma will likely drive the development of novel antiplatelet therapies for the prevention or management of CNS disorders. Healthy platelet-derived preparations are also gaining increasing recognition as a potential neuroprotective biotherapy. Although further preclinical and clinical investigation is required, the possibility of administering well-characterized and standardized platelet lysates to the brain may offer an affordable treatment for CNS disorders. The potential of platelet EVs as a standalone biotherapy is another area where preclinical studies are urgently required. Both therapies could be made readily available worldwide using the domestic blood supply.
Acknowledgments
The authors would like to thank Ms. Rowan Tweedale and Dr. Odette Leiter for helpful comments on the manuscript and Mr. David Brici for assistance with the preparation of the figures and graphical abstract.
Funding support for this article was provided by the Ministry of Science and Technology (MOST) (110-2314-B-038-079), Brazil Family Foundation for Neurology, and Donald and Joan Wilson Foundation.
Authorship
Contribution: T.B. and T.L.W. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Thierry Burnouf, Graduate Institute of Biomedical Materials and Tissue Engineering College of Biomedical Engineering Taipei Medical University 250 Wuxing St Xinyi District, Taipei City 110 11031, Taiwan; e-mail: thierry@tmu.edu.tw; and Tara Walker, Queensland Brain Institute, Building 79, Upland Rd, The University of Queensland, St Lucia, Brisbane, QLD 4072, Australia; e-mail: t.walker1@uq.edu.au.