TO THE EDITOR:
DNMT3A overgrowth syndrome (DOS, also known as Tatton-Brown-Rahman Syndrome [TBRS]) is an overgrowth syndrome caused by de novo germline mutations in the gene encoding the de novo DNA-methyltransferase 3A (DNMT3A). The original report of DOS included 13 affected individuals, none with a history of cancer.1 Mutations were found in all 3 functional domains of DNMT3A and likely caused loss-of-function of the mutant allele.1-4 Affected individuals shared distinctive facial features, intellectual disability, obesity, and tall stature. Although somatic mutations in DNMT3A are among the most common initiating events for patients with normal karyotype acute myeloid leukemia (AML) and clonal hematopoiesis,5-9 mutations of DNMT3A are rarely found in pediatric patients with AML.10
The natural history of DOS has not yet been fully described. According to the “Tatton-Brown-Rahman Syndrome Community,” there are ∼200 known affected individuals worldwide (Jill Kiernan, “TBRS Community,” personal communication, 4 October 2021). A recent study evaluating the effects of germline DNMT3A pathogenic variants on hematopoiesis found similar total white cell counts and hemoglobin levels in DOS patients and controls, but noted some patients with DOS had an increased fraction of circulating neutrophils, a decrease in B cells, and mild macrocytosis.4 We recently described the DNA methylation landscape for 11 patients with DOS (including one with a history of AML) compared with unaffected controls, which showed focal hypomethylation in nonleukemic peripheral blood cells that was more pronounced in patients with DNMT3AR882H than other DNMT3A mutations.3 Although reports of AML have been described in 2 patients with DOS,2,11 and a single T-lymphoblastic lymphoma/leukemia,12 the incidence of hematopoietic malignancies in patients with DOS is not yet clear.
We identified 8 patients with DOS with a history of a hematopoietic malignancy (Table 1), and their case presentations are summarized in the supplemental Appendix, available on the Blood Web site. Myeloid and lymphoid malignancies were found, with a median age at diagnosis of 9.6 years, in an equal number of males and females. DNMT3A mutations identified in these patients were scattered across the gene (with the most common mutation site at amino acid R882), and the majority are listed in the COSMIC database as being associated with hematologic malignancies (Table 1).
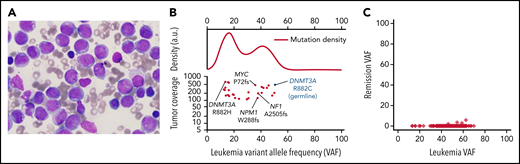
Case 1 developed AML with an FAB M4 phenotype at age 12 (Figure 1A). We were able to obtain samples to perform exome or whole-genome sequencing (WGS) on presentation and remission samples (and a buccal swab sample) from this case (case 3 in Smith et al3) to identify cooperating mutations in the AML sample (Figure 1B; supplemental Table 1). The founding clone at presentation contained AML-associated mutations in NPM1 (VAF 37.6%), NF12505fs (VAF 37.9%), and MYCP72PT (VAF 35.9%); all the somatic founding clone mutations were completely cleared in a peripheral blood sample obtained 4.4 years later, when the patient was in morphologic remission (Figure 1C). At presentation, the patient had a second DNMT3AR882 mutation (R882H) that was either in a subclone or in an independent nonmalignant clone (ie, clonal hematopoiesis). This mutation was not detected in the remission sample. WGS revealed no structural variants. In the AML case reported by Hollink et al,11 cytogenetics revealed a complex karyotype, and the AML sample had an AML-associated mutation (PTPN11T73I). Tumor samples were not available for study in the other 6 cases. The study of additional AMLs should clarify whether patients with DOS have cooperating mutations common in adult DNMT3A-driven AMLs (ie, NPMc, FLT3-ITD, IDH1/2, PTPN11, and others5,10), and whether there are genotype-phenotype correlations depending on the location of the specific DNMT3A mutation in each patient with DOS.
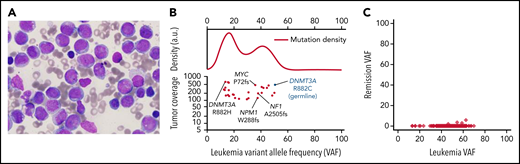
Case 1, AML characterization. (A) Presentation bone marrow aspirate, AML blasts with folded nuclei, prominent nucleoli, and fine chromatin, consistent with FAB subtype M4. The stain is Wright-Giemsa; original magnification is ×1000. (B) Variant allele frequencies (VAFs) of high-coverage-depth (>100×) somatic mutations from the patient's bone marrow plotted against tumor coverage. The kernel density plot (top; a.u., arbitrary units) is suggestive of at least 1 major subclonal population. AML-related mutations are labeled (bottom). The germline DNMT3A mutation is denoted in blue for context. (C) VAFs of the leukemia and remission samples, showing that all leukemic cells were cleared. Only 0.03% of remission sample reads at mutation locations had variant-supporting reads, well within the error rate of the sequencing process.
Case 1, AML characterization. (A) Presentation bone marrow aspirate, AML blasts with folded nuclei, prominent nucleoli, and fine chromatin, consistent with FAB subtype M4. The stain is Wright-Giemsa; original magnification is ×1000. (B) Variant allele frequencies (VAFs) of high-coverage-depth (>100×) somatic mutations from the patient's bone marrow plotted against tumor coverage. The kernel density plot (top; a.u., arbitrary units) is suggestive of at least 1 major subclonal population. AML-related mutations are labeled (bottom). The germline DNMT3A mutation is denoted in blue for context. (C) VAFs of the leukemia and remission samples, showing that all leukemic cells were cleared. Only 0.03% of remission sample reads at mutation locations had variant-supporting reads, well within the error rate of the sequencing process.
We also identified 2 patients in the TARGET database of pediatric AML whose WGS revealed germline DNMT3A variants (PARBTV: DNMT3AR882H, and PARZIA: DNMT3AN501S). Because we have limited information about their clinical phenotypes, they cannot be assigned definitively as patients with DOS. Details of these cases, including potential cooperating mutations (IDH2R172K for "PARBTV" and +10 for "PARZIA"), are presented in the supplemental Appendix and supplemental Tables 4 and 5. Similarly, 2 cases of germline DNMT3AP709A mutations in a mother and son were associated with the development of AML (with an acquired mutation or loss of heterozygosity in the residual wild-type DNMT3A allele, and an associated IDH2R172K mutation) at ages 50 and 30, respectively,13 but neither had the syndromic features of DOS and cannot be formally included in this collection (supplemental Table 3).
The incidence of DOS is likely to be underestimated given the novelty of the diagnosis and lack of testing in underserved areas. Of the 202 cases known to the “TBRS Community,” the maximum age is 39 years (mean, 14 years); the majority of cases are from North America (92/202, 45%) and Europe (89/202, 44%), with similar prevalence in males and females (112/199 male, 56%; Jill Kiernan, TBRS Community, personal communication, 4 October 2021). Prospective monitoring of these individuals will be required to determine whether the development of cancer continues to increase with age or peaks during childhood. With those caveats, we found 8 patients with DOS with hematopoietic malignancies out of these 202 known cases, for an estimated prevalence of ∼4%. This is similar to the incidence of leukemia seen in a recent report of patients with TP53 mutations (Li-Fraumeni syndrome) with leukemia presenting between ages 6 and 35.13 The risk of leukemia and lymphoma in childhood (0 to 14 years) is 42 cases per million.14 In the adolescent and young adult age group (15 to 39 years), the incidence is 108 cases per million.15 Combined, the risk is therefore of ∼150 per million for ages 0 to 39, suggesting that there may be a >250-fold increase in the risk of hematologic malignancy in patients with DOS.
Although most childhood-onset hematologic malignancies are due to sporadic mutations, multiple predisposition syndromes have been described, mainly for leukemia.16-18 New predisposition syndromes have been recently discovered, and as our ability to test for genomic alterations in patient populations improves, the list will likely continue to grow. Given the data presented here, and the finding of spontaneous leukemias developing in germline mouse models of DOS,3,19 we suggest that DOS should be classified as a hematologic malignancy predisposition syndrome.
We suggest that annual monitoring of the complete blood counts of patients with DOS should be performed on a research basis to better understand the natural history of these individuals, and their risk of developing hematopoietic malignancies. If abnormalities are noted, the complete blood counts should be repeated in 2 to 4 weeks. If abnormalities persist or worsen, the patient should undergo bone marrow evaluation, including cytogenetic analysis and next-generation sequencing panel sequencing for adult AML-associated gene mutations. In 2017, a consensus statement from a panel of pediatric hematologists-oncologists, geneticists, and genetic counselors was published to give guidance on surveillance of children with leukemia predisposition syndromes.20-22 One of the key recommendations was the need for education of at-risk patients and their families about the signs and symptoms of leukemia that would prompt evaluation (fever, fatigue, easy bruising and petechiae, pallor, splenomegaly, and lymphadenopathy).
Of note, there have also been 3 reports of central and peripheral nervous system tumors in young patients with DOS (medulloblastoma, pituitary adenoma, and ganglioneuroma; case 3 of this series).12,21,22 These data suggest that DOS patients may also be at increased risk for central nervous system and peripheral nervous system tumors. As more patients with DOS are identified, it will be important to enroll them into a registry that records all documented cancers with tissue of origin and associated mutation data, and to monitor them carefully for the development of cancer, to better define the natural history of this syndrome (https://tbrsregistry.iamrare.org).
Acknowledgments
The authors thank the Tatton-Brown-Rahman Syndrome (TBRS) Community, and especially Jill Kiernan and Kerry Grens, for help in identifying the cases, and the patients and families themselves for their participation. The authors also thank all referring physicians for providing records and obtaining blood and bone marrow samples for the study.
This work was supported by an Alex’s Lemonade Stand Foundation Young Investigator Award (M.A.F.), the National Institutes of Health (NIH), Eunice Kennedy Shriver National Institute of Child Health and Human Development grant 5K12HD07622408 (M.A.F.), ASH Fellow to Faculty Award (A.M.S.), NIH, National Cancer Institute grants R35 CA220500 (J.M.M.), NCI R50 CA211782 (Miller), PO1CA101937, and NCI R35CA197561, and the Barnes Jewish Hospital Foundation (T.J.L.).
Authorship
Contribution: M.A.F., A.M.S., and T.J.L. designed the research; S.E.H., E.J.D., M.O., D.F., R.W., S.D., J.M.M., A.F.R., M.D.W., F.C., R.B.F., S.K., and M.S. provided clinical advice/services; M.A.F., C.A.M., D.H.S., and T.J.L. analyzed the data; and M.A.F. and T.J.L. wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Timothy J. Ley, Washington University School of Medicine, 660 South Euclid Ave, Box 8007, St Louis, MO 63110; e-mail: timley@wustl.edu.
Data may be found at the National Institutes of Health, National Center for Biotechnology Information Genomics of Acute Myeloid Leukemia, dbGap phs000159 v.11.
The online version of this article contains a data supplement.