Key Points
WHIM patients treated orally once daily with 400 mg of mavorixafor had increased total white blood cell, neutrophil, and lymphocyte counts.
WHIM patients treated with mavorixafor for ≥6 months showed reduced annualized infection rates and decreased wart numbers.

Abstract
Warts, hypogammaglobulinemia, infections, and myelokathexis (WHIM) syndrome is a rare primary immunodeficiency caused by gain-of-function mutations in the CXCR4 gene. We report the safety, tolerability, pharmacokinetics, pharmacodynamics, and preliminary efficacy of mavorixafor from a phase 2 open-label dose-escalation and extension study in 8 adult patients with genetically confirmed WHIM syndrome. Mavorixafor is an oral small molecule selective antagonist of the CXCR4 receptor that increases mobilization and trafficking of white blood cells from the bone marrow. Patients received escalating doses of mavorixafor, up to 400 mg once daily. Five patients continued on the extension study for up to 28.6 months. Mavorixafor was well tolerated with no treatment-related serious adverse events. At a median follow-up of 16.5 months, we observed dose-dependent increases in absolute neutrophil count (ANC) and absolute lymphocyte count (ALC). At doses ≥300 mg/d, ANC was maintained at >500 cells per microliter for a median of 12.6 hours, and ALC was maintained at >1000 cells per microliter for up to 16.9 hours. Continued follow-up on the extension study resulted in a yearly infection rate that decreased from 4.63 events (95% confidence interval, 3.3-6.3) in the 12 months prior to the trial to 2.27 events (95% confidence interval, 1.4-3.5) for patients on effective doses. We observed an average 75% reduction in the number of cutaneous warts. This study demonstrates that mavorixafor, 400 mg once daily, mobilizes neutrophil and lymphocytes in adult patients with WHIM syndrome and provides preliminary evidence of clinical benefit for patients on long-term therapy. The trial was registered at www.clinicaltrials.gov as #NCT03005327.
Introduction
CXCR4 and its ligand CXCL12 regulate the trafficking of white blood cells (WBCs) between the bone marrow and blood compartments.1 Constitutively increased CXCR4/CXCL12 signaling causes warts, hypogammaglobulinemia, infections, and myelokathexis (WHIM) syndrome, a rare combined primary immunodeficiency with severe neutropenia (Online Mendelian Inheritance in Man Entry #193670) that is caused by C-terminal autosomal-dominant gain-of-function mutations of the CXCR4 gene2-4 (Figure 1A).
![Schematic overview of mavorixafor in WHIM syndrome. (A) Gain of function mutations in the CXCR4 receptor. Heterozygous autosomal dominant gain-of-function CXCR4 mutations cause WHIM syndrome. Schematic diagram of the human CXCR4 receptor showing extracellular, transmembrane, and intracellular domains and known mutated C-terminal (COOH) residues reported to cause WHIM syndrome. CXCR4 mutations truncate the C terminus by premature termination (red) or frameshift (gray). A stop mutation and a single amino acid substitution that causes WHIM syndrome are reported in position 343 (red and blue). (B) Chemical structure of mavorixafor (X4P-001). (S)-N1-((1H-benzo[d]imidazol-2-yl)methyl)-N1-(5,6,7,8-tetrahydroquinolin-8-yl)butane-1,4-diamine. (C) Study flowchart. Patient P5 discontinued early because of a psoriasiform rash. Patients P3 and P4 did not continue on the extension phase because of personal preference and trial fatigue. (D) Mean (+ standard error) plasma concentration vs time profile of mavorixafor by dose presented on a semilog scale. The horizontal dotted line represents the IC50 of mavorixafor for CXCR4 inhibition, corrected for total (bound and unbound) drug levels.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/136/26/10.1182_blood.2020007197/1/m_bloodbld2020007197f1.png?Expires=1769081043&Signature=UsfB35v~Y8xxDZGqw-6-Xs-GGmpuDxKnH80qUN~vcxeEK727VcBguHP3TEoDfUfrMu0nFV8fSCTqqNmIUoNdCjUqoohQCW-FhWiJCVyrfeRerIsxgrHP0tTTsR~Sf~D1EAWIKiv6D60zzKmzLTJZy19FXoVgKMH-tMO2l9xwWT-BEk3QJJgisCWHEqe9ECLCc0fUL5DU13Oci164GeAeogn1FvxTXgXNGEbvRbIi1zLv8dgjj87Me~W-G5oixzPog5~4y6Laseg44xoRCRBYddFr8YldMlLYAdhYhEHDHR0GhhaWHkAh2ms1UqOftGawBT99kHMIDsWyNO1SuzThgg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Schematic overview of mavorixafor in WHIM syndrome. (A) Gain of function mutations in the CXCR4 receptor. Heterozygous autosomal dominant gain-of-function CXCR4 mutations cause WHIM syndrome. Schematic diagram of the human CXCR4 receptor showing extracellular, transmembrane, and intracellular domains and known mutated C-terminal (COOH) residues reported to cause WHIM syndrome. CXCR4 mutations truncate the C terminus by premature termination (red) or frameshift (gray). A stop mutation and a single amino acid substitution that causes WHIM syndrome are reported in position 343 (red and blue). (B) Chemical structure of mavorixafor (X4P-001). (S)-N1-((1H-benzo[d]imidazol-2-yl)methyl)-N1-(5,6,7,8-tetrahydroquinolin-8-yl)butane-1,4-diamine. (C) Study flowchart. Patient P5 discontinued early because of a psoriasiform rash. Patients P3 and P4 did not continue on the extension phase because of personal preference and trial fatigue. (D) Mean (+ standard error) plasma concentration vs time profile of mavorixafor by dose presented on a semilog scale. The horizontal dotted line represents the IC50 of mavorixafor for CXCR4 inhibition, corrected for total (bound and unbound) drug levels.
Schematic overview of mavorixafor in WHIM syndrome. (A) Gain of function mutations in the CXCR4 receptor. Heterozygous autosomal dominant gain-of-function CXCR4 mutations cause WHIM syndrome. Schematic diagram of the human CXCR4 receptor showing extracellular, transmembrane, and intracellular domains and known mutated C-terminal (COOH) residues reported to cause WHIM syndrome. CXCR4 mutations truncate the C terminus by premature termination (red) or frameshift (gray). A stop mutation and a single amino acid substitution that causes WHIM syndrome are reported in position 343 (red and blue). (B) Chemical structure of mavorixafor (X4P-001). (S)-N1-((1H-benzo[d]imidazol-2-yl)methyl)-N1-(5,6,7,8-tetrahydroquinolin-8-yl)butane-1,4-diamine. (C) Study flowchart. Patient P5 discontinued early because of a psoriasiform rash. Patients P3 and P4 did not continue on the extension phase because of personal preference and trial fatigue. (D) Mean (+ standard error) plasma concentration vs time profile of mavorixafor by dose presented on a semilog scale. The horizontal dotted line represents the IC50 of mavorixafor for CXCR4 inhibition, corrected for total (bound and unbound) drug levels.
The acronym “WHIM” highlights 4 manifestations of the syndrome5 : warts, hypogammaglobulinemia, infections, and myelokathexis, the retention of mature neutrophils in the bone marrow.2,3 The laboratory manifestations include leukopenia, severe neutropenia, profound lymphopenia, and monocytopenia that result in recurrent bacterial infections and unusual susceptibility to certain viral infections, which have been linked to a higher incidence of cancer.6,7 Bacterial infections frequently present as pneumonia, recurrent otitis, sinusitis, or cellulitis and are associated with chronic morbidity,6-8 such as bronchiectasis or hearing loss. Selective immunodeficiency to human papilloma virus (HPV) results in treatment-refractory mucocutaneous warts that are at high risk for malignant transformation,7,9 and Epstein-Barr virus infections may be linked to a higher risk for lymphoma,10 further increasing the risk of malignancy in WHIM syndrome.
Mavorixafor is a selective oral small molecule allosteric antagonist of the CXCR4 receptor11 that is in development for the treatment of WHIM syndrome (Figure 1B). Herein, we present the clinical and hematological results of a dose-finding and long-term extension study of 8 patients affected with genetically confirmed WHIM syndrome treated for up to 28.6 months.
Methods
Design
X4P-001-MKKA is an open label prospective international dose-escalation study of mavorixafor in adult patients with WHIM syndrome followed by an extension study conducted in 2 clinical trial sites located in Australia and the United States.
Patients aged ≥18 years with a pathogenic CXCR4 mutation6 and absolute neutrophil count (ANC) ≤400 per microliter and/or an absolute lymphocyte count (ALC) ≤650 per microliter were included. Patients treated with plerixafor within the prior 2 months and/or granulocyte colony-stimulating factor (G-CSF) and/or taking any medications with potential drug interactions modulated via cytochrome P450 or P-glycoprotein within the prior 2 weeks were excluded. Highly effective contraception was a requirement for patients and their partners.
Dose escalation from 50-400 mg once daily was based on the threshold-adjusted area under the curve for ANC (AUCANC) and the threshold-adjusted area under the curve for ALC (AUCALC), with thresholds of 600 cells per microliter for ANC and 1000 cells per microliter for ALC over 24 hours. Patients were dose escalated if AUCANC <2000 or AUCALC <5000 hours × cells/μL. Dose escalation was at the discretion of the sponsor, with the agreement of the investigator(s), based on AUCANC and AUCALC values. Patients who completed ≥25 weeks of the initial escalation could elect to continue escalation up to 400 mg once daily or extend treatment at the minimum effective dose with biannual assessments.
Study X4P-001-MKKA was conducted in accordance with Good Clinical Practice and the principles of the Declaration of Helsinki, following a written protocol approved by the institutional review boards or ethics committee for each center. All patients provided written informed consent.
Assessments and outcomes
The primary objective was to evaluate the safety and tolerability of the drug and the dose required to achieve a consistent increase in circulating neutrophils and lymphocytes. Exploratory objectives evaluated the efficacy of long-term mavorixafor on infection rate, skin warts, WBC count, ANC, ALC, and absolute monocyte count (AMC).
The primary end point, used to determine dose escalation, was the mean value of the threshold-adjusted AUCANC and/or AUCALC collected over 24 hours (with ANC threshold of 600 cells per microliter and ALC threshold of 1000 cells per microliter). To calculate the AUC above the threshold for each visit, the area below the threshold was subtracted from the total AUC (supplemental Figure 1, available on the Blood Web site). Exploratory efficacy end points were: (1) absolute and fold change from baseline for total WBC, ANC, ALC, and AMC, (2) annualized infection rate at each dose level compared with the prestudy prior 12 months (supplemental Figure 2), and (3) number of warts on the hands and feet compared with baseline. In addition, a post hoc analysis examined the time, in hours, during which ANC was maintained at >500 cells per microliter or ALC was maintained at >1000 cells per microliter, defined as the time above threshold for ANC (TATANC) and time above threshold for ALC (TATALC), respectively.
Safety and tolerability in the safety population were assessed according to the National Cancer Institute Common Terminology Criteria for Adverse Events (version 4.03 or higher) and the Medical Dictionary for Regulatory Activities (version 19.0). All adverse events were investigator assessed and monitored until resolution.
Pharmacokinetics (PK) and pharmacodynamics (PD) samples were obtained in the safety population during 24-hour in-residence periods (at weeks 5, 13, and 21 and during the extension period) at the following time points: predose; 30, 60, and 90 minutes; and 2, 3, 4, 8, 12, 16, and 24 hours. Mavorixafor plasma concentrations were analyzed using a validated reverse-phase high performance liquid chromatography with tandem mass spectrometry detection. PK parameters, such as area under the plasma concentration-time curve for the 24-h dosing interval (AUC0-24h), maximum observed plasma concentration (Cmax), and time to maximum plasma concentration (Tmax), were determined by noncompartmental analysis (using Phoenix WinNonlin 8.1) of the steady-state plasma concentration vs time from last dose before sample collection for each subject.
Statistical analysis
The data cutoff date for this analysis was 14 June 2019. Descriptive statistics summarized patient demographics, clinical characteristics, PK and PD response, and safety parameters. If a subject had multiple PK assessments at the same dose, the median PK parameters were calculated for that subject at that dose before deriving the summary statistics. All data processing and statistical analyses were done using SAS version 9.4 or higher. Infection events were summarized using annualized rate, and the 95% confidence interval (CI) was calculated based on Poisson distribution. The infection rate in the 12 months prior to the study was based on the review of patients’ medical records (supplemental Figure 2). Dermatological response evaluated the number of warts on hands and feet.
Results
Patient baseline characteristics
We enrolled 8 patients (P1-P8) for up to a maximum duration of 28.6 months. Patient demographics, baseline hematological and clinical characteristics are summarized in Table 1. The mean age at inclusion was 33 years (range, 18-57). Sanger sequencing identified pathogenic gain-of-function CXCR4 mutations in all patients: R334X (6/8), E343X (1/8), and S324Pfs365X (1/8) (Figure 1A). All patients had a history of profound leukopenia, neutropenia, lymphopenia, and monocytopenia and exhibited ≥2 characteristic disease manifestations, including warts and/or hypogammaglobulinemia, and infections requiring antibiotic therapy within the past 12 months (Table 1). Infection sequelae included bronchiectasis (n = 1) and perforated tympanic membranes or marked scarring (n = 5) that resulted in severe hearing loss in 2 patients. Patient P2 had a history of squamous cell carcinoma on the arm, basal cell carcinoma on the neck, and atypical cervical squamous cells on a Papanicolaou test. Prior treatments included short-term plerixafor (2/8 patients), and all patients (8/8) had received various regimens of prophylactic G-CSF). No prophylactic antibiotic regimen was prescribed during the trial. Patient P8 received chronic prophylactic immunoglobulin substitution in the 12 months prior to, as well as during, the study. This patient was excluded from the PD response analysis because of high WBC counts in a context of chronic bronchitis and splenectomy for cholelithiasis.
Disposition and exposure
During the study, 8 patients received mavorixafor for a median of 16.5 months (mean, 15.4 months; range, 6 days to 28.6 months). Five patients continued on the long-term extension study (Figure 1C) for a median of 19.6 months (mean, 21.2 months; range, 16.3-28.6). Compliance was high (0.9% of missed doses based on pill count). Two patients each started with 50, 100, 200, or 300 mg (supplemental Table 1). Seven patients received mavorixafor, 300 mg once daily; 3 escalated to 400 mg once daily (Table 2; supplemental Figure 3) based on AUCANC. Total median drug dose exposure for all 8 patients was 151 200 mg (range, 1200-268 950). Patients P3 and P4 exited the study after 6 months. Patient P5, who had previously discontinued a trial investigating low-dose plerixafor because of a psoriasiform rash developed a similar rash after 6 days on mavorixafor and discontinued (Figure 1C; Table 1). At the date of submission of this manuscript, 5 patients remain on the extension study.
Treatment-emergent adverse events
Seven patients (87.5%) experienced ≥1 treatment emergent adverse event (TEAE). Three patients experienced 11 grade 1 related TEAEs: nausea (4 events), nasal dryness (2 events), dry mouth (2 events), dyspepsia (1 event), conjunctivitis (1 event), and dermatitis psoriasiform rash (1 event). TEAE frequency did not increase with dose. No pregnancies were recorded. Infections were the most common type of TEAE, were considered unrelated to the study drug, and were reported as outcomes (yearly infection rate). There were 2 unrelated grade 3 TEAEs (cholecystitis in a patient with multiple gallstones and procedural pain). No related serious adverse event or significant findings in clinical chemistry or urinalysis, ophthalmology, or electrocardiograms was reported.
Mavorixafor, 400 mg once daily, was established as a therapeutically effective dose
Patients received escalated doses of mavorixafor based on mean AUCANC and AUCALC: 50 mg (n = 2), 100 mg (n = 4), 150 mg (n = 2), 200 mg (n = 3), 300 mg (n = 7), and 400 mg (n = 3). Not all patients received all doses. Plasma concentration vs time profile is presented in Figure 1D. PK parameters are shown in Table 2 and supplemental Table 3. Mavorixafor was rapidly absorbed with a Tmax of 1.25 to 2 hours postdose. Based on the power model test for dose proportionality,12 the Cmax increased almost proportionally with dose, whereas AUC0-24h increased more than proportionally. The steady-state Cmax and AUC0-24h for the 300- and 400-mg once-daily doses were similar (2430 and 2260 ng/mL for Cmax and 9500 and 8060 hours × ng/mL for AUC0-24h, respectively), but this may be an artifact of the small number of subjects (n = 3) in the 400-mg dose group or interoccasion (within-subject) PK variability due to limited sampling occasions.
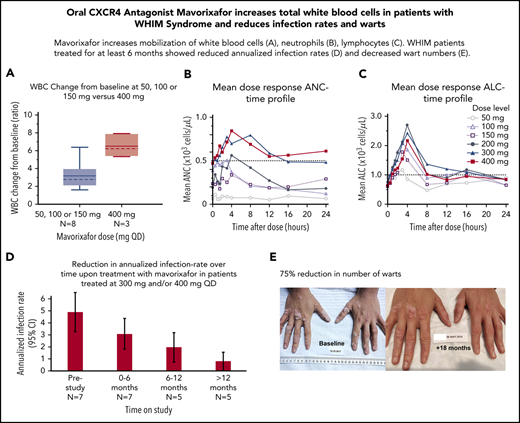
We observed a dose-dependent increase in the mean WBC, ANC, ALC, and AMC (Figures 2A-C, 4), as well as the AUCANC (Figure 2D; supplemental Table 2) and AUCALC (supplemental Table 2). When AUC0-24h was plotted against TATANC and TATALC, we observed a strong correlation with ANC (correlation coefficient, r = 0.73) and a weak correlation with ALC (r = 0.22) (Figure 3A-B). Similar relationships were seen with mavorixafor Cmax (data not shown). For patients treated with 300 mg or 400 mg, the median (range) TATANC was 12.6 (0-24.0) hours (n = 7) compared with 1.9 (0-7.3) hours (n = 4) for doses ≤150 mg, representing a 6.6-fold increase; the median TATALC was 16.9 (9.6-24.0) hours. Median TATANC at 300 mg was 8.6 (0.0-24.0) hours (n = 7). Median TATANC at 400 mg was 13.8 (1.7-24.0) hours (n = 3). Two patients treated with 300 mg increased their mean AUCANC above 2000 hours × cells/μL and did not receive higher doses (Figure 4); TATANC for these 2 patients was 22.5 and 24.0 hours. Of the 3 patients escalated to 400 mg, mean AUCANC was >2000 hours × cells/μL and TATANC was >12 hours (ie, 13.6 and 24 hours) in 2 patients (Figure 2A; Table 2; supplemental Table 3). Based on these observations, and particularly the increase in WBC at higher doses, which indicates the mobilization of multiple leukocyte subsets, the 400-mg once-daily dose was determined to be therapeutically effective.
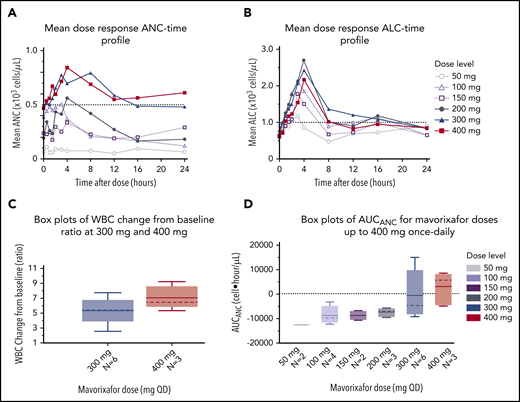
Global hematological response to mavorixafor. (A) Mean dose response ANC-time profile. The ANC threshold (500 neutrophils per microliter) is indicated by the dashed line. (B) Mean dose response ALC-time profile. The ALC threshold (1000 cells per microliter) is indicated by the dashed line. (C) Increasing concentrations of mavorixafor result in an increase in the total WBC counts in patients with WHIM syndrome. Box plots show WBC change from baseline ratio at 4 hours for 300-mg and 400-mg once-daily mavorixafor. For box plots, the dashed line is the median; the solid line is the arithmetic mean; the lower and upper ends of the boxes are the 25th and 75th percentiles, respectively; the lower whiskers show the lowest data value still within 1.5 interquartile range (IQR) of the lower quartile, and the upper whiskers show the highest value still within 1.5 IQR of the upper quartile. (D) Box plots of AUCANC for mavorixafor doses up to 400 mg, once daily. Box plots show AUCANC >600 cells per microliter threshold over 24 hours. The dotted line represents a null net AUC relative to the ANC theshold of 600 neutrophils/μL. The values above the dotted line represents the net AUC result above this threshold of 600 neutrophils/μL. N, number of patients; QD, once daily.
Global hematological response to mavorixafor. (A) Mean dose response ANC-time profile. The ANC threshold (500 neutrophils per microliter) is indicated by the dashed line. (B) Mean dose response ALC-time profile. The ALC threshold (1000 cells per microliter) is indicated by the dashed line. (C) Increasing concentrations of mavorixafor result in an increase in the total WBC counts in patients with WHIM syndrome. Box plots show WBC change from baseline ratio at 4 hours for 300-mg and 400-mg once-daily mavorixafor. For box plots, the dashed line is the median; the solid line is the arithmetic mean; the lower and upper ends of the boxes are the 25th and 75th percentiles, respectively; the lower whiskers show the lowest data value still within 1.5 interquartile range (IQR) of the lower quartile, and the upper whiskers show the highest value still within 1.5 IQR of the upper quartile. (D) Box plots of AUCANC for mavorixafor doses up to 400 mg, once daily. Box plots show AUCANC >600 cells per microliter threshold over 24 hours. The dotted line represents a null net AUC relative to the ANC theshold of 600 neutrophils/μL. The values above the dotted line represents the net AUC result above this threshold of 600 neutrophils/μL. N, number of patients; QD, once daily.

Dose-dependent hematologic improvement. Mavorixafor produces an initial peak elevation in WBC count, ALC, ANC, and AMC at ∼4 hours postdose. The ALC threshold of 1000 cells per microliter and the ANC threshold of 500 neutrophils per microliter are indicated by dashed lines.
Dose-dependent hematologic improvement. Mavorixafor produces an initial peak elevation in WBC count, ALC, ANC, and AMC at ∼4 hours postdose. The ALC threshold of 1000 cells per microliter and the ANC threshold of 500 neutrophils per microliter are indicated by dashed lines.
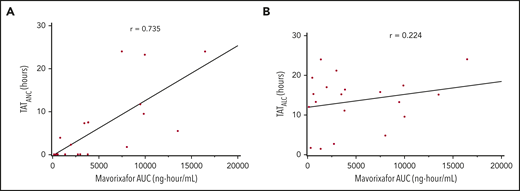
Correlation between the hematologic response and mavorixafor concentration. The correlation coefficient is represented by r. Subjects may contribute to 1 or more time points because of dose escalation and/or repeated sampling over time. (A) Correlation between TATANC and mavorixafor concentration. (B) Correlation between TATALC and mavorixafor concentration.
Correlation between the hematologic response and mavorixafor concentration. The correlation coefficient is represented by r. Subjects may contribute to 1 or more time points because of dose escalation and/or repeated sampling over time. (A) Correlation between TATANC and mavorixafor concentration. (B) Correlation between TATALC and mavorixafor concentration.
Reduction in the annualized infection rate
The retrospective yearly infection rate in the 12 months prior to the trial was 4.63 (95% CI, 3.3-6.3) events (n = 8) in the safety population (Figure 5A), which was comparable to 5 (95% CI, 3.34-6.66) yearly events for the 7 patients treated with up to 300 mg (Figure 5B; supplemental Table 3) and to 4.53 (95% CI, 2.1-9.5) yearly events in 4 patients treated with subtherapeutic doses of 50 mg, 100 mg, or 150 mg. In contrast, we observed a decreased yearly infection rate of 2.41 (95% CI, 1.29-4.48) for the 7 patients treated at 300 mg once daily and 2.14 (95% CI, 1.11-4.10) for 400 mg once daily. No patient required G-CSF for infection events or prophylactic antibiotics on study. At 300 mg and/or 400 mg on the extension study, the infection frequency was correlated to the time on treatment. Indeed, during the first 6 months, the yearly infection rate was 3.14 (95% CI, 1.83-4.46) (n = 7) compared with 2.0 (95% CI, 0.76-3.24) for patients treated for 6 to 12 months (n = 5) and 0.8 (95% CI, 0.02-1.58) for patients treated >12 months (n = 5), providing preliminary evidence that long-term treatment may further prevent infections in this population (Figure 5B; supplemental Table 4). Overall, during the 12 months prior to the study, the most common types of infections involved the lower respiratory tract, followed by the upper respiratory tract and skin. This contrasted with the observed infections on study; the most common infections concerned the upper respiratory tract, followed by the lower respiratory tract.

Clinical response to treatment. (A) Reduction in the annualized infection rate upon treatment with mavorixafor, 300 mg and 400 mg once daily. Reduction in the annualized infection rate upon treatment with mavorixafor, 300 mg and 400 mg once daily, compared with the 12 months prior and to lower doses of mavorixafor (50 to 150 mg once daily). The retrospective yearly infection rate in the 12 months prior to the trial was calculated based on the safety population (n = 8). The yearly infection rate is derived from the sum of the number of infection events for each patient divided by the total exposure time (in years). (B) Reduction in the annualized infection rate over time upon treatment with mavorixafor in patients treated at 300 mg and/or 400 mg once daily. Yearly infection rates compared with the 12 months prior to the study. The retrospective yearly infection rate in the 12 months prior to the trial was calculated based on the patients treated up to ≥300 mg (n = 7). The yearly infection rate at each treatment period is derived from the sum of the number of infection events for each patient divided by the exposure time (in years) at each time period. (C-D) Significant reduction in the number of cutaneous warts during long-term once-daily mavorixafor treatment. Patient P6 was treated with increasing doses of mavorixafor alone for a total of 18 months (including 14 months at 400 mg once daily). The patient was not given imiquimod or other treatments for HPV. (C) Warts at baseline. (D) Warts 18 months later, after 14 months at 400 mg. A significant decrease in wart burden could be seen after 6 months on treatment.
Clinical response to treatment. (A) Reduction in the annualized infection rate upon treatment with mavorixafor, 300 mg and 400 mg once daily. Reduction in the annualized infection rate upon treatment with mavorixafor, 300 mg and 400 mg once daily, compared with the 12 months prior and to lower doses of mavorixafor (50 to 150 mg once daily). The retrospective yearly infection rate in the 12 months prior to the trial was calculated based on the safety population (n = 8). The yearly infection rate is derived from the sum of the number of infection events for each patient divided by the total exposure time (in years). (B) Reduction in the annualized infection rate over time upon treatment with mavorixafor in patients treated at 300 mg and/or 400 mg once daily. Yearly infection rates compared with the 12 months prior to the study. The retrospective yearly infection rate in the 12 months prior to the trial was calculated based on the patients treated up to ≥300 mg (n = 7). The yearly infection rate at each treatment period is derived from the sum of the number of infection events for each patient divided by the exposure time (in years) at each time period. (C-D) Significant reduction in the number of cutaneous warts during long-term once-daily mavorixafor treatment. Patient P6 was treated with increasing doses of mavorixafor alone for a total of 18 months (including 14 months at 400 mg once daily). The patient was not given imiquimod or other treatments for HPV. (C) Warts at baseline. (D) Warts 18 months later, after 14 months at 400 mg. A significant decrease in wart burden could be seen after 6 months on treatment.
We analyzed the clinical response of patient P8, who presented elevated baseline ANCs due to chronic bronchitis with a history of lung exacerbations and bronchiectasis. This patient continued immunoglobulin prophylaxis on study at the same dose and regimen as in the 12 months prior. The infection rate of 8 events in the 12 months prior to the study decreased to 2 infections in the first 6 months at 300 mg once daily, then to 1 infection in the following 6 months, and no infections in the most recent 12 to 16.5 months. Patient P2 demonstrated increased WBC counts, ALC, and AMC at 400 mg, but only modestly increased ANC (Figure 4), and nevertheless had a yearly infection rate that decreased from 5 events in the year preceding the trial to 2.13 annualized events at 400 mg once daily.
Reduction in number of cutaneous warts after treatment with mavorixafor
Patients (n = 4) with ≥1 cutaneous wart on their hands and/or feet at baseline and treated with up to 300 mg or 400 mg once daily, demonstrated an average 75% reduction in the number of warts after 5 to 18 months on study, without the use of topical imiquimod or other treatment. The number of warts was further reduced in the 2 patients who remained on study beyond 12 months, with an average 80% reduction. On treatment, larger lesions evolved into multiple smaller warts, and continued treatment allowed clearance of most lesions. Patient P6 displayed the most severe treatment-refractory warts, with 174 lesions on the hands and feet at baseline. Over the course of the study, we observed a gradual, but marked, disappearance of the warts. After 6 months, a >50% decrease in the number of warts (69 lesions) was observed. At the most recent examination after 18 months on study (and 14 months on mavorixafor, 400 mg once daily), the patient presented 33 lesions, corresponding to a >80% reduction in wart burden. This is documented photographically (Figure 5C-D).
Discussion
In this study, we present the first safety, hematological, and clinical efficacy results of a dose-finding PK and long-term extension study of 8 patients affected with genetically confirmed WHIM syndrome treated with once-daily oral CXCR4 antagonist mavorixafor for up to 28.6 months.
There is no approved therapy targeting the molecular mechanism of WHIM syndrome. WHIM syndrome has been recognized as an example of a severely debilitating or life-threatening hematologic disorder by the US Food and Drug Administration.13 Therapeutic options include G-CSF to correct the neutropenia, immunoglobulin replacement to increase immunoglobulin G (IgG) levels, and antibiotics to treat acute bacterial episodes or prevent infections. Unfortunately, evidence of efficacy using these treatments in WHIM patients is lacking.8 Additionally, warts remain refractory to treatment,8 and no treatment addresses the associated lymphopenia or monocytopenia. Although proof-of-concept clinical studies using low-dose twice-daily subcutaneous injections of plerixafor (AMD3100, Mozobil), a CXCR4 antagonist, were shown to safely and durably correct panleukopenia in WHIM syndrome,9,14-17 mavorixafor is the only orally bioavailable CXCR4 antagonist.
Mavorixafor is a small molecule selective allosteric antagonist of the CXCR4 receptor that increases mobilization and trafficking of WBCs from the bone marrow.18,19 Mavorixafor binds to the extracellular region of the CXCR4 receptor11,18 and specifically and reversibly inhibits receptor signaling of the most common pathogenic CXCR4 variants (R334X and E343X) and wild-type CXCR4 receptors.18 In vitro data show that mavorixafor acts as an allosteric inhibitor that blocks CXCR4 binding with a 50% inhibition concentration (IC50) of 12.5 nM (4.4 ng/mL), with high selectivity against other chemokine (CXC and CC motifs) receptors.18 The mean concentrations of mavorixafor in the plasma after the 400 mg once-daily dose remained above the corresponding IC50 value (corrected for total bound and unbound drug) for CXCR4 inhibition, as shown by the dotted line in Figure 1D, for the full 24 hours of the dosing interval.
In this study, the favorable tolerability profile is highlighted by the high compliance with treatment and by the fact that 5 of the 8 patients enrolled remain on the long-term extension. Furthermore, patients and healthy volunteers in 9 clinical studies (oncology,20-23 n = 99; healthy volunteers,19,24,25 n = 70; HIV,26,27 n = 16; and WHIM syndrome; n = 8) have received mavorixafor from 5 to 1064 days (mean, 221 days) (supplemental Table 5); overall, it was well tolerated, with no mavorixafor-related serious adverse event grade 3 or higher.
The patients in this study were representative of WHIM patients in the literature. Genetically, 6 presented the most common pathogenic R334X variant reported to cause WHIM syndrome,16 and 2 carried other pathogenic variants that result in a premature translational stop signal in the receptor (E343X and S324Pfs365X).4,14 Clinically, the heterogeneous presentation reported in this study is consistent with the literature. This phenotypic variability may lead to diagnostic delay and underestimation of the disease prevalence.6,28
The study determined that a therapeutic dose to sustainably achieve an increase in neutrophils >500 cells per microliter and lymphocytes >1000 cells per microliter for >12 hours was 400 mg once daily; patients maintained on prolonged therapy showed a reduction in the yearly infection rate and a reduction in the number of cutaneous warts.
Dose escalation was based on AUCANC and AUCALC collected over 24 hours (with ANC threshold of 600 cells per microliter and ALC threshold of 1000 cells per microliter). However, the high variability in the AUCANC and AUCALC limited the clinical relevance of these measures. Therefore, we examined, as a post hoc analysis, a more clinically relevant end point: TATANC and TATALC. The 500 cells per microliter ANC threshold is used to predict the risk of serious bacterial infections in patients who have neutropenia resulting from disorders of bone marrow production.29 With this analysis, we observed that 4 of the 6 evaluable patients increased their TATANC, at 300 mg and/or 400 mg once daily, to >12 hours, with higher WBC cells, increased ALC and AMC, and a reduction in the infection rate compared with the 12 months prior to study initiation. We now suggest that TATANC is an objective and consistent biomarker of the response to therapy in WHIM patients. We further propose that TATANC is a valid PD end point that reflects global immunological improvement and leukocyte mobilization and correlates with plasma drug exposures (Figure 3A) and clinical end points (Figure 5). TATANC is the primary end point in our current phase 3 study in WHIM syndrome.
This study demonstrated that treatment duration at effective doses correlated with reduction in infection rate (Figure 5B). The yearly infection rate for patients treated with 400 mg once daily decreased to 2.14 (95% CI, 1.11-4.10) events compared with the 4.63 (95% CI, 3.3-6.3) events in the prior 12 months (Figure 5A). This was correlated with increased ANC and ALC (Figure 4; Table 2; supplemental Table 3), which translated as increased total WBC count ratios from baseline (Figure 2C). Prolonged treatment duration at the effective doses of 300 mg and/or 400 mg once daily showed decreasing yearly infection rate trends. During the first 6 months, the yearly infection rate was 3.14 (95% CI, 1.83-4.46) events (n = 7); the infection rate decreased to 2.0 (95% CI, 0.76-3.24) events between 6 and 12 months (n = 5) and to 0.8 (95% CI, 0.02-1.58) events for patients treated >12 months (n = 5), providing preliminary evidence of the importance of long-term treatment to prevent infections and their related morbidity in WHIM syndrome (Figure 5B).
In this study, we have shown a correlation between increases in ANC and improved clinical outcomes for patients; in certain cases, clinical benefit has also been observed in patients who did not achieve target ANC thresholds, as is the case for patient P2. This may be because mavorixafor treatment facilitated and hastened the neutrophil response to infections, thereby reducing the infection rate. However, the study design does not allow us to determine whether this occurred. Although patient P2 did not sustain ANC >500 cells per microliter threshold at the 400-mg dose, an approximately threefold increase in neutrophils and lymphocytes, compared with values at the 50-mg dose, were observed based on 4-hour postdose values. In addition, the ability of mavorixafor to increase multiple WBC subsets distinguishes mavorixafor from currently available therapies that only increase ANC. Finally, the pan-elevation of WBCs in response to mavorixafor, as a result of improved leukocyte trafficking, may also contribute to an overall improved immunological function and may further decrease the risk of infection in WHIM patients.
Prolonged oral CXCR4 inhibition showed an impact on the HPV-induced skin warts. The precise immune mechanisms of HPV immunity are not completely understood in WHIM syndrome.30 One hypothesis is that the control of HPV may involve myeloid and lymphoid cells, including plasmacytoid dendritic cells, which express CXCR4 and are deficient in the blood of untreated WHIM patients.31 Further understanding of the importance of the myeloid compartment in the control of HPV can be gained from the report of 1 WHIM patient whose warts were cleared after a spontaneous cure by chromothripsis,32 which resulted in a correction of the myeloid lineage in the bone marrow and supports the contribution of this lineage to HPV immunity. In the literature, WHIM syndrome patients treated with long-term twice-daily subcutaneous injections of plerixafor showed partial responses in the appearance of cutaneous warts,6,15,16 and HPV genotyping found that, after 7 to 18 months on treatment, the γ HPV predominance decreased.33 These observations may account for the reduction in skin warts in response to CXCR4 antagonism, because of the ability of this treatment to mobilize the myeloid and lymphoid compartments from the marrow.
We observed a median 75% reduction in the number of cutaneous warts on the hands and feet in the 4 patients with warts at baseline who were treated with mavorixafor, up to 300 mg or 400 mg, once daily. Treatment benefit was even more apparent in the patients who remained on the extension treatment. In particular, patient P6, who has a history of severe treatment-resistant warts, showed 80% clearance of the lesions after 18 months on treatment (14 months on 400 mg, once daily), without any adjunct therapy for warts (Figure 5C-D); reductions > 50% in wart burden were evident after 6 months on study. One of the shortcomings of the wart assessment in this study is that response to treatment was based only on counting the number of warts on the hands and feet, whereas size or severity of lesions was not recorded. In addition, larger lesions evolved into multiple smaller warts, so counting the number of warts potentially underestimated clinical improvement. Furthermore, the impact of mavorixafor on genital warts was not evaluated. Future trials should characterize the dermatological response in WHIM by assessing the severity and global change over time of cutaneous warts in association with standardized photography procedures and systematic HPV genotyping. The evaluation of anogenital warts may provide additional insight into the long-term impact on malignant transformation risk.
In summary, these data highlight that mavorixafor provides improved and durable clinical efficacy compared with current therapeutic options for WHIM syndrome. The excellent tolerability profile and therapeutic benefit of long-term mavorixafor on infection rate and wart burden are being investigated further in the ongoing phase 3 study of mavorixafor, once daily, in patients with WHIM syndrome.
Data sharing requests should be sent to David Dale (dcdale@uw.edu).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank all of the patients who participated in this study, as well as Raj Bhardwaj for contributing to PK analysis and Xia Luo for assisting with figures.
This work was supported by research funding from X4 Pharmaceuticals, Inc.
Authorship
Contribution: K.J.G. and T.E. designed the study; D.C.D., F.F., and A.A.B. managed the study, with support and input from M.K. and V.M.; S.B.C., D.C.D., A.A.B., F.F., V.G., W.T., H.J., and R.S. analyzed and interpreted data; and S.B.C. and D.C.D. wrote the first draft of the manuscript, which was reviewed, modified, and approved by D.C.D., A.A.B., F.F., K.J.G., M.K., V.M., V.G., W.T., H.J., R.S., and S.B.C.
Conflict-of-interest disclosure: D.C.D. and F.F. received research funding for the trial from X4 Pharmaceuticals, Inc. D.C.D. serves on the advisory board for X4 Pharmaceuticals, Inc. T.E., V.G., W.T., H.J., R.S., and S.B.C. are employees in X4 Pharmaceuticals, Inc and may own equity in the company. K.J.G. is a consultant for X4 Pharmaceuticals, Inc and is affiliated with Zymo Consulting Group LLC. The remaining authors declare no competing financial interests.
Correspondence: David C. Dale, Department of Medicine, University of Washington, 1959 NE Pacific St, Room AA522, Box 356422, Seattle, WA 98195; e-mail: dcdale@uw.edu.