In this issue of Blood, demonstrate that active endoplasmic reticulum–associated degradation (ERAD) of misfolded proteins is required to maintain hematopoietic stem cell (HSC) quiescence and, hence, proper long-term blood formation.1 They identify a mechanism by which the Sel1L protein, a key member of the ERAD complex, maintains HSCs in a state of low mTORC1 activity, thereby preventing HSC proliferation. These data implicate that steady-state protein quality control directly contributes to keeping HSCs outside of the cell cycle.
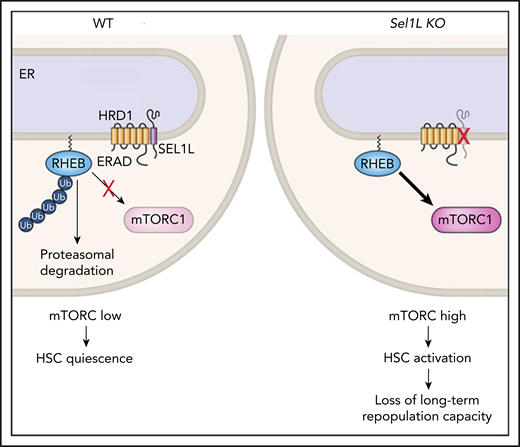
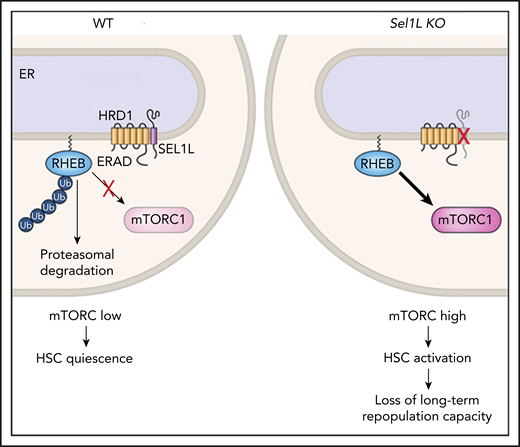
RHEB is normally maintained at low levels in HSCs via HRD1/SEL1L (ERAD), which targets it for proteasomal degradation, thereby keeping mTORC activation low. When Sel1L is genetically deleted, RHEB is stabilized and mTORC1 is activated, leading to HSC loss of quiescence. ER, endoplasmic reticulum; KO, knockout; WT, wild-type.
RHEB is normally maintained at low levels in HSCs via HRD1/SEL1L (ERAD), which targets it for proteasomal degradation, thereby keeping mTORC activation low. When Sel1L is genetically deleted, RHEB is stabilized and mTORC1 is activated, leading to HSC loss of quiescence. ER, endoplasmic reticulum; KO, knockout; WT, wild-type.
HSCs have immense regenerative potential, because 1 cell can reconstitute the whole blood system for an entire lifetime. Counterintuitively, lifelong blood production can only be achieved if cells divide very infrequently and otherwise reside in a specific state outside of the cell cycle, termed quiescence. Effectively, the most quiescent HSCs serve as a reservoir that only contributes to blood production under severe stress. Perturbation of the quiescence to division balance almost inevitably leads to loss of long-term HSC function and is of high clinical relevance in blood disorders ranging from cytopenias to malignancies. Quiescence is an actively maintained state of low metabolism with specific hallmarks shared by all quiescent cells, ranging from bacteria and yeast to mammalian adult tissue stem cells, including HSCs.2 These hallmarks encompass a preference for glycolytic metabolism, in addition to low levels of transcription, ribosome biogenesis, and protein synthesis. Given HSCs’ impressive longevity, it is no surprise that strict quality-control mechanisms, such as enhanced autophagy and specific DNA damage responses, are also essential to maintain optimal genetic integrity and fitness of quiescent HSCs.3
Once a protein is synthesized, protein folding occurs in the endoplasmic reticulum and is a naturally error-prone process that often results in the generation of misfolded or unfolded proteins. Failure to correctly manage unfolded proteins can culminate in cell death. External stress conditions, such as oxidative stress, can increase protein misfolding, activating the unfolded protein response (UPR) that relieves endoplasmic reticulum stress via numerous routes, including activation of the Sel1L/Hrd1 ERAD complex, which is known to target misfolded proteins to proteasomal degradation. Previous work from the Liu group, as well as others, has demonstrated that an intact UPR pathway is very important to safeguard HSC identity and proper function under stress conditions.4-6
Here, the investigators set out to determine whether protein quality control via ERAD needs to remain engaged during quiescence, despite the fact that cells have overall low protein synthesis. Their first important finding is that the HSCs that divide the least (as measured by label-retaining assays) have lower levels of protein aggregates and higher levels of Sel1L messenger RNA than do HSCs with a higher divisional history. Second, deletion of Sel1L via 2 distinct methods in mice led to a reduction in HSC frequency in the bone marrow and loss of repopulation capacity upon transplantation. Sel1L-knockout HSCs displayed an “activated” phenotype, with an increased proportion entering the cell cycle, increased cell size, and mTOR activation. The investigators then quite elegantly identify Rheb, a regulator of mTOR, as a new protein substrate of the ERAD complex, which fails to be ubiquitinated and degraded by the proteasome in the absence of Sel1L. Thus, Sel1L deletion leads to high levels of mTOR (see figure). Importantly, inhibition of mTOR via rapamycin or genetic means rescued HSC numbers and repopulation capacity close to those of wild-type mice.
Altogether, this study demonstrates that ERAD-mediated protein quality-control mechanisms are essential in HSCs with low protein synthesis to prevent mTOR upregulation and excessive activation. Importantly, Liu et al provide yet another confirmation that the less frequently an HSC divides, the more reliant it is on quality-control mechanisms and that these same mechanisms reinforce quiescence. In work published concomitantly with theirs, Xu et al also found that Sel1L deletion leads to loss of repopulating HSCs.7 Focusing on the fact that improper quality control of cell surface proteins may be highly deleterious to HSCs, they identified Mpl, the receptor of thrombopoietin, as a target protein of the ERAD complex. Sel1L-knockout HSCs accumulated aggregates of misfolded Mpl intracellularly, displayed decreased levels of functional Mpl on their surface, and could not be retained in their perivascular niche. The mechanistic insights provided by the 2 groups are highly synergistic because complex interactions with the niche, as well as thrombopoietin itself,8,9 are critical to maintain HSC quiescence. In fact, it is highly likely that ERAD provides folding control for many more molecules that contribute to HSC function. Thus, future studies will have to examine the physiological range of ERAD activity in the hematopoietic system over a lifetime, especially with aging and following infection or chronic inflammation. Mutations also often change the probability that certain proteins will be misfolded. As such, it would not be surprising if perturbations in proteostatic control pathways contributed to HSC clonal expansions, as well as the development and progression of malignancies.
Conflict-of-interest disclosure: The author declares no competing financial interests.