

Key Points
In patients with lymphadenopathy and/or splenomegaly with elevated DNTs, ALPS must be suspected; genetics and biomarkers can confirm this.

Abstract
Autoimmune lymphoproliferative syndrome (ALPS) is a rare immunodeficiency caused by mutations in genes affecting the extrinsic apoptotic pathway (FAS, FASL, CASP10). This study evaluated the clinical manifestations, laboratory findings, and molecular genetic results of 215 patients referred as possibly having ALPS. Double-negative T-cell (DNT) percentage and in vitro apoptosis functional tests were evaluated by fluorescence-activated cell sorting; interleukin 10 (IL-10) and IL-18 and soluble FAS ligand (sFASL) were measured by enzyme-linked immunosorbent assay. Genetic analysis was performed by next-generation sequencing. Clinical background data were collected from patients’ records. Patients were categorized into definite, suspected, or unlikely ALPS groups, and laboratory parameters were compared among these groups. Of 215 patients, 38 met the criteria for definite ALPS and 17 for suspected ALPS. The definite and suspected ALPS patient populations showed higher DNT percentages than unlikely ALPS and had higher rates of lymphoproliferation. Definite ALPS patients had a significantly more abnormal in vitro apoptosis function, with lower annexin, than patients with suspected ALPS (P = .002) and patients not meeting ALPS criteria (P < .001). The combination of elevated DNTs and an abnormal in vitro apoptosis functional test was the most useful in identifying all types of ALPS patients; the combination of an abnormal in vitro apoptosis functional test and elevated sFASLs was a predictive marker for ALPS-FAS group identification. Lymphoproliferation, apoptosis functional test, and DNTs are the most sensitive markers; elevated IL-10 and IL-18 are additional indicators for ALPS. The combination of elevated sFASLs and abnormal apoptosis function was the most valuable prognosticator for patients with FAS mutations.
Introduction
Autoimmune lymphoproliferative syndrome (ALPS; also called Canale-Smith syndrome) is a rare immunodeficiency with several pathognomonic hallmarks, such as nonmalignant chronic lymphoproliferation, unexplained lymphadenopathy, splenomegaly, immune-mediated cytopenia, and hypergammaglobulinemia caused by an abnormal extrinsic FAS-mediated apoptotic pathway.1-3 Defective lymphocyte apoptosis could principally cause autoimmune manifestations, mainly multilineage cytopenias and less frequently nephritis, hepatitis, uveitis, arthritis, or colitis. Furthermore, patients with ALPS as well as genetically affected siblings without an ALPS phenotype are predisposed to several malignancies, such as solid tumors (eg, thyroid, breast, and liver) and leukemia; the risk of non-Hodgkin lymphoma is up to 50 times greater compared with the general population.4-6
In ALPS, the defective extrinsic apoptotic signaling is caused by germline and somatic mutations in FAS (TNFRSF6, CD95, AP01), FASLG, and CASP10 genes, encoding Fas cell surface death receptor protein, FAS ligand (FASL) protein, and caspase 10.7 A majority of ALPS patients present with germline heterozygous or somatically acquired FAS mutations. Rarely, FAS mutations are inherited in an autosomal recessive manner.2,3,8,9 The FAS gene encodes a protein receptor expressed on B and T lymphocyte lineages. FASLG gene mutations are identified in extremely rare cases of ALPS.10 On the basis of the underlying pathogenic genetic variants, ALPS is classified into 4 categories according to the affected genes, including ALPS-FAS, ALPS-FASLG, and ALPS-CASP102,7 ; in ALPS patients without a defined mutation in these genes (20% to 30% of cases), the condition is classified as ALPS-U (unknown).2,6,8,11-17 Patients with pathogenic caspase-8 (CASP8) gene variants resulting in caspase-8 deficiency are classified as having an ALPS-related disorder characterized by defective T-, B-, and NK-cell activation in addition to defective FAS-induced lymphocyte apoptosis.2,10,18
Impaired activation–induced cell death in patients with ALPS results in the development of an abnormal T-cell repertoire.19 The accumulation of an autoreactive CD3+TCRαβ+ CD4−CD8− double-negative T-cell (DNT) population was initially reported in 1992 as a main characteristic of ALPS. This cell population normally accounts for ∼1% of all T-cell subsets in the paracortical region and peripheral blood (PB) of healthy individuals.20,21 The DNTs resemble normally differentiated T cells and usually express naïve T-cell markers, such as CD45RA, on their cell surface. However, these cells are highly proliferative and are able to induce upregulation in mammalian target of rapamycin and several other growth pathways.15,22 A skewed T- helper cell profile and reduced number of CD27+ memory B cells have been described. Other consequential laboratory features of ALPS include raised levels of soluble FASL (sFASL), interleukin-10 (IL-10), and IL-18.
This retrospective study aimed to review the ALPS 2010 diagnostic criteria3 and define the most predictive and useful biomarkers and their combinations by evaluating 215 patients with clinical evidence of ALPS, including information on clinical manifestation, laboratory findings, and molecular genetic results.
Methods
Cohort description
Between 2008 and 2018, blood samples from 215 patients (132 males and 83 females; median age, 12.3 years; range, 1 month to 76 years) with a clinical suspicion of ALPS were referred to the Immunology Laboratory of the Great Ormond Street Hospital (London, United Kingdom). Patients belonged to 197 unrelated families. Clinical data included information about the presence of chronic nonmalignant, noninfectious lymphadenopathy and/or splenomegaly, cytopenia, and immunoglobulin G (IgG) and vitamin B12 levels. The Immunology Laboratory assessed the DNTs, performed a lymphocyte apoptosis function assay, and measured sFASL, IL-10, and IL-18. Genetic analysis was performed in the North East Thames Regional Genetics Laboratory (London, United Kingdom). Clinical, immunological, and genetic laboratory data were collected retrospectively, and available clinical outcome data were also included. This study was approved by the Bloomsberry Research Ethics Committee and was conducted in accordance with the Declaration of Helsinki.
Assessment of laboratory parameters
The measurement of CD3+TCRαβ+ CD4−CD8− DNTs was performed by flow cytometry. The pathogenic level of DNTs has been defined as >1.8% of all α/β T cells based on the gating strategy of the laboratory.2,23
The in vitro FAS-mediated apoptosis assay used PB mononuclear cells stimulated for 6 to 7 days with anti-CD3 and IL-2. After incubation, cells were stimulated with anti-FAS antibody to trigger apoptosis. Apoptosis can be detected by the surface staining of cell membrane proteins (annexin V). Apoptotic cells positive for annexin V but negative for the DNA viability stain (7AAD) were enumerated by flow cytometry. In normal individuals, the difference between the anti-FAS antibody–stimulated sample and –unstimulated sample was >3.0-fold change.23 Annexin V expression after anti-FAS antibody treatment was usually at a low level in ALPS patients. The assessment of healthy unrelated control samples was undertaken, with each patient sample serving as a quality control for the variables that may have affected this functional assay (eg, transport, temperature, reagents). The normal annexin V fold change was defined as >3.0, defective apoptosis was defined as <2.0-fold change, and the range between 2.0 and 3.0 was described as equivocal.2,3
The Human IL-10 Quantikine ELISA Kit (D1000B), Human IL-18 Quantikine ELISA Kit (DL180), and Human FAS Ligand/TNFSF6 Quantikine ELISA Kit (DFL00B) were used from R&D Systems. The normal ranges were defined as <40, <500, and <200 pg/mL, respectively. The normal range of vitamin B12 was between 228 and 1510 pg/mL, and the normal range of IgG was within 5.4 to 16.1 g/L.
Genetic studies
Genes affecting the extrinsic apoptotic pathway (FAS, FASLG, CASP10) and an additional 79 genes included in the targeted immunodeficiency and gastrointestinal enrichment panel were analyzed by next-generation sequencing (NGS).24 The library preparation was performed using a Sure Select XT custom kit followed by sequencing on Illumina MiSeq. Detected variants were evaluated based on the recommendations of the American College of Medical Genetics and Genomics (ACMG).25 Web-based VarSome helped with variant annotation and classification.26 Potential splice variants were evaluated using Human Splicing Finder software.27 Identified variants were confirmed by Sanger sequencing.
Applied diagnostic evaluation
The clinical symptoms and laboratory biomarkers were evaluated by the ALPS 2010 diagnostic protocol,3 which defines 4 major criteria, including: chronic nonmalignant lymphoproliferation (>6 months) with splenomegaly and/or lymphadenopathy, DNT elevation (1.8%) in PB, defective in vitro FAS-mediated apoptosis, and recognizable genetic pathogenic mutation (in FAS, FASLG, and CASP10 genes). Along with these major criteria, there are 6 minor criteria, including: multilineage cytopenia, elevated levels of IgG, IL-10, IL-18, or vitamin B12 in serum, and increased sFASL level in plasma. At least 3 major criteria or 2 major and 2 minor criteria are required for a diagnosis of definite ALPS. The diagnostic protocol was updated according to the working definitions for clinical diagnosis of primary immunodeficiencies of the European Society for Immunodeficiencies,28 where elevated DNT ratio was only a major, but not a required, criterion. The diagnostic protocol was extended; suspected ALPS was defined as the fulfillment of 2 major and 1 minor criteria. This group was created to include those patients who had possibly received immunosuppressive treatments at the time of referral. Cases lacking information about at least 2 major diagnostic and at least 1 minor criterion were defined as nonevaluable.
Statistical analysis
For the analysis of continuous variables, the Shapiro-Wilk test was used to test for normality. None of the parameters examined showed normal distribution; thus, the Kruskal-Wallis or Mann-Whitney U test was performed, with Dunn’s test for post hoc analysis. For the analysis of categorical variables, we applied Pearson’s χ2 test. Data were analyzed using IBM SPSS Statistics 23, Prism GraphPad, and Microsoft Excel.
Results
Clinical and laboratory features
Of the 215 patients referred with clinical evidence of ALPS, clinical background was recorded for 87 patients, including lymphoproliferation, lymphadenopathy, and splenomegaly, and for 101 patients, hemoglobin, platelet, and white blood cell counts were available. DNT information was available for 146 cases, an apoptosis functional test was performed for 192, and genetic results were accessible for 86 (Figure 1). In addition, IgG (n = 66), vitamin B12 (n = 31), sFASL (n = 126), and IL-10 and IL-18 (n = 30) were also measured. Patients were categorized as nonevaluable in 75 cases. From the evaluable patient group, 38 patients (27.1%) met the criteria for definite ALPS (median age at first referral, 9.3 years; range, 4 months to 77 years; supplemental Figure 1, available on the Blood Web site). Another 17 patients (12.1%) fulfilled the criteria for suspected ALPS (median age at first referral, 13.1 years; range, 1 month to 19 years; supplemental Figure 1), whereas 85 patients (60.7%) did not meet the ALPS diagnostic criteria (unlikely ALPS group; median age at first referral, 10.3 years; range, 2 months to 64 years).
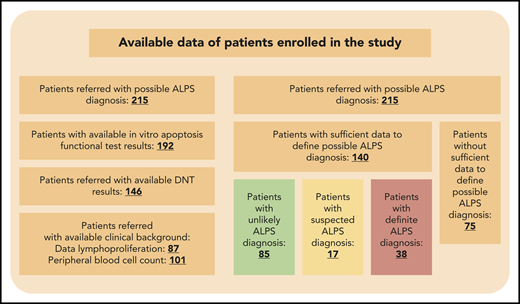
Patients enrolled in the study with the accessible diagnostic results. A total of 215 patients were referred with potential ALPS diagnosis. The minimal criteria for evaluation were at least 2 major criteria and 1 minor criterion; 140 patients were evaluable. Patients were considered as definite ALPS patients if at least 3 major or at least 2 major and 2 minor criteria were observed. Patients were considered suspected ALPS patients if at least 2 major criteria and 1 minor criterion were observed.
Patients enrolled in the study with the accessible diagnostic results. A total of 215 patients were referred with potential ALPS diagnosis. The minimal criteria for evaluation were at least 2 major criteria and 1 minor criterion; 140 patients were evaluable. Patients were considered as definite ALPS patients if at least 3 major or at least 2 major and 2 minor criteria were observed. Patients were considered suspected ALPS patients if at least 2 major criteria and 1 minor criterion were observed.
The clinical criteria for ALPS, including chronic lymphoproliferation with or without lymphadenopathy/splenomegaly, were met more frequently in definite (n = 33 [97.1%] of 34) and suspected (n = 14 [87.5%] of 16) ALPS groups than in the unlikely ALPS group (n = 22 [55%] of 40; P < .0001). Multilineage cytopenia was observed in 69% (n = 20 of 29) of definite, 56.3% (n = 9 of 16) of suspected, and 56.6% (n = 30 of 53) of unlikely ALPS populations (P = .5168; Figure 2A). Outcome data were available in a limited number of patients. Of 34 definite ALPS patients, 3 (n = 3 [8.82%] of 34), along with another 3 suspected ALPS patients (n = 3 [19.75%] of 16), underwent splenectomy. Lymphoma development was described in 2 (n = 2 [5.88%] of 34) definite ALPS patients.
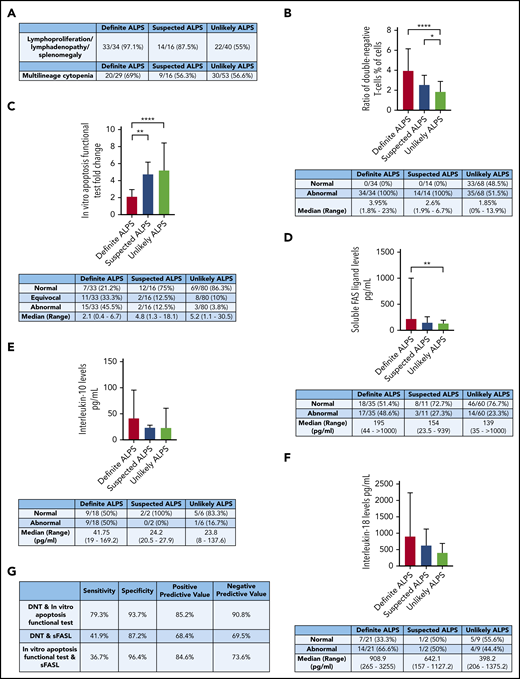
Laboratory markers and clinical parameters of the cohort. (A) Clinical data of the enrolled patients: lymphoproliferation (P < .0001 definite vs unlikely ALPS) and multilineage cytopenia (P = .5168). (B) DNTs. (C) The in vitro apoptosis functional test. (D) The level of sFASL (P = .0674 definite vs suspected ALPS). (E) IL-10 (P = .0624 definite vs suspected and unlikely ALPS). (F) IL-18 (P = .0615 definite vs suspected and unlikely ALPS). (G) Assessment of the marker combination comparing the definite ALPS and unlikely ALPS groups. All data are presented as median ± interquartile range. *P = .0496 (suspected vs unlikely ALPS), **P = .0019 (definite vs suspected ALPS; panel C), **P = .0013 (definite vs unlikely ALPS; panel D), ****P < .0001 (definite vs suspected ALPS; panel B), ****P < .0001 (definite vs unlikely ALPS; panel C).
Laboratory markers and clinical parameters of the cohort. (A) Clinical data of the enrolled patients: lymphoproliferation (P < .0001 definite vs unlikely ALPS) and multilineage cytopenia (P = .5168). (B) DNTs. (C) The in vitro apoptosis functional test. (D) The level of sFASL (P = .0674 definite vs suspected ALPS). (E) IL-10 (P = .0624 definite vs suspected and unlikely ALPS). (F) IL-18 (P = .0615 definite vs suspected and unlikely ALPS). (G) Assessment of the marker combination comparing the definite ALPS and unlikely ALPS groups. All data are presented as median ± interquartile range. *P = .0496 (suspected vs unlikely ALPS), **P = .0019 (definite vs suspected ALPS; panel C), **P = .0013 (definite vs unlikely ALPS; panel D), ****P < .0001 (definite vs suspected ALPS; panel B), ****P < .0001 (definite vs unlikely ALPS; panel C).
Abnormally high DNTs were observed in all definite (n = 34) and all suspected ALPS patients (n = 14). In the unlikely ALPS population, 35 (51.5%) of 68 cases showed DNT elevation. The median DNT ratio was 3.95% (range, 1.8% to 23.0%) in definite, 2.6% (range, 1.9% to 6.7%) in suspected, and 1.85% (range, 0.0% to 13.9%) in unlikely ALPS groups. There was significant difference between definite ALPS and unlikely ALPS groups (P < .0001) and between suspected ALPS and unlikely ALPS groups (P = .0496; Figure 2B).
In the definite ALPS group (n = 33), 15 patients had an abnormal apoptosis functional test (45.5%), 11 had an equivocal result (33.3%), and 7 had a normal result; in the suspected ALPS group (n = 16), 2 patients (12.5%) had abnormal, 2 (12.5%) had equivocal, and 12 had normal apoptosis function. In patients with an unlikely ALPS diagnosis (n = 80), 3 and 8 (3.8 and 10%) had an abnormal or equivocal apoptosis functional test result, respectively. In the definite ALPS group, the median annexin expression change was 2.1-fold (range, 0.4- to 6.7-fold). In suspected ALPS patients, the median was 4.8-fold (range, 1.3- to 18.1-fold), and in the unlikely ALPS group, the median was 5.2-fold (range, 1.1- to 30.5-fold; P = .0019 between definite and suspected ALPS groups; P < .0001 between definite and unlikely ALPS groups). However, a significant difference was not shown between the suspected and unlikely ALPS groups (P = .8953; Figure 2C).
A significant difference in sFASL levels was seen between the definite and unlikely ALPS groups (P = .0013; definite ALPS: median, 195 pg/mL; range, 44 to >1000 pg/mL; suspected ALPS: median, 154 pg/mL; range, 23.5-939 pg/mL; unlikely ALPS: median, 139 pg/mL; range, 35 to >1000 pg/mL; Figure 2D). IL-10 levels showed a tendency toward being elevated in definite ALPS (median, 41.8 pg/mL; range, 19-169.2 pg/mL) compared with suspected ALPS (median, 24.2 pg/mL; range, 20.5-27.9 pg/mL) and unlikely ALPS patients (median, 23.8 pg/mL; range, 8-137.6 pg/mL; P = .0624; Figure 2E). Additionally, a tendency toward increased IL-18 levels was also observed between the groups (definite ALPS: median, 909 pg/mL; range, 265-3255 pg/mL vs suspected ALPS: median, 642 pg/mL; range, 157-1127 pg/mL and unlikely ALPS: median, 398 pg/mL; range, 206-1375 pg/mL; P = .0615; Figure 2F).
Description of biomarker combinations
Optimal biomarker combinations were tested by setting up groups of 2 where both markers were positive or either of them was negative; the marker combinations were compared between definite and unlikely ALPS groups (Figure 2G). The combination of DNTs and abnormal in vitro apoptosis functional test was positive in 79.3% (23 of 29) in definite ALPS patients and negative in 93.7% (59 of 63) of unlikely ALPS patients; therefore, the sensitivity of this combination was 79.3%, and the specificity was 93.7%. The DNT and sFASL combination was positive in 41.9% (13 of 31) of definite ALPS patients and negative in 87.2% (41 of 47) of unlikely ALPS patients; therefore, the sensitivity was 41.9%, and the specificity was 87.2%. The in vitro apoptosis functional test and sFASL combination was positive in 36.7% (11 of 30) of definite ALPS and negative in 96.4% (53 of 55) of unlikely ALPS patients; therefore, the sensitivity of this combination was 36.7%, and the specificity was 96.4%. All 3 combinations showed a significant difference between the definite and unlikely ALPS groups (P < .0001 for DNT and in vitro apoptosis functional test; P = .0040 for DNT and sFASL; P = .00012 for in vitro apoptosis functional test and sFASL).
Genetic results
Genetic results were available for 87 patients. We considered pathogenic variants, likely pathogenic variants, and variants of unknown significance (VUSs), classified according to the ACMG guidelines as genetic variants that possibly alter function (Table 1). Variants with higher populational frequencies than expected for ALPS occurrence, along with variants that do not alter protein function (intronic not affecting splice site, exonic synonymous, or neutral/tolerated by in silico prediction, such as Provean, SIFT, and MetaSVM), were referred to as likely benign/benign genetic variants. Previous clinical and in vitro functional studies considering gene variants were reviewed.
FAS gene variants were identified in 21 patients, 17 of whom were considered as having a potentially functional variant (14 definite and 1 suspected ALPS). The remaining 2 cases did not have enough clinical or laboratory data for evaluation (1 individual was identified during family screenings of affected proband). Two frameshift and 2 nonsense pathogenic variants were identified in heterozygous forms in 4 definite ALPS patients. Three of these loss-of-function mutations had not been reported before in the literature (c.715_721delGTCATGA, p.Val239HisfsTer2; c.719_722delinsAGTTA, p.Met240LysfsTer7; and c.76C>T, p.Gln26Ter), whereas 1 of the nonsense variants had been described (c.219C>A, p.Cys73Ter).4,29 Regarding likely pathogenic missense FAS variants, 3 novel (c.742T>G, Phe248Val; c.794A>G, Asp265Gl; and c.826C>A, Gln276Lys) and 2 previously reported (c.749G>A, Arg250Gln and c.776T>G, Ile259Arg)30,31 were identified. Interestingly, 2 patients with previously described likely pathogenic missense mutations showed a normal in vitro FAS-mediated apoptosis assay. The intronic c.335-9_335-6delATTT variant was categorized as a VUS (not identified in the general population; in ALPS patients, its intronic location does not affect the conserved splice region). The variant lies in close proximity to an acceptor splice site; the in silico splice prediction suggested that the variant altered the splice site, most probably affecting splicing.27
The following benign FAS gene variants were identified. The FAS c.136A>C (p.Thr46Pro) was categorized as a VUS according to ACMG criteria. The variant was found in a 2-year-old patient not showing the characteristic biomarker positivity of ALPS, which reduced, but did not exclude, the pathogenic role of this genetic variant in ALPS. Further development of ALPS manifestations later in life cannot be excluded. One FAS synonymous variant (c.642T>C) and 5 FAS intronic variants were found in 3 patients, but minor allele frequencies of these single-nucleotide polymorphisms are much higher in the normal population than the expected disease frequency of ALPS (the synonymous variant also cooccurred with a variant previously reported as pathogenic in CASP10). Somatic FAS mutations were not detected in our evaluable patient group with sufficient clinical data. This may reflect a lack of sensitivity of the sequencing assay, because genetic analysis was not performed on sorted DNTs.
In the pathogenic FAS variant patient group, 14 patients had abnormal DNT levels, and for 3 patients, results were not available. Nine patients had an abnormal, 2 had an equivocal, and 3 had a normal in vitro lymphocyte apoptosis functional test. In the other 3 cases, no results were available.
CASP10 variants were found in 5 patients (2 definite, 1 suspected, and 2 nonevaluable). The CASP10 variant (c.1216A>T; p.Ile406Leu) has been previously published as pathogenic in ALPS; in vitro functional studies have proven its pathogenicity, although its MAF (0.4%) and in silico prediction programs are defined as benign variant (ACMG categorization: likely benign).32 CASP10 p.Ile406Leu occurred in 2 probands (with definite and suspected ALPS diagnoses) and 2 family members, who were nonevaluable. CASP10 c.295A>G (p.Lys99Glu) was found in 1 patient with definite ALPS. The variant had not been previously reported in association with ALPS and has low MAF in the general population (0.04%). Because the in silico analyses were inconclusive, ACMG classification was likely benign. CASP10 c.1228G>A (p.Val410Ile) was found in 1 patient and cooccurred with another missense CASP10 variant (c.1216A>T) in our cohort, supporting its prediction as a benign variant.28,33,34 ALPS accompanied by possibly pathogenic or benign CASP10 variants had an elevated percentage of DNTs (n = 2) but a normal in vitro lymphocyte apoptosis functional test and sFASL. The clinical manifestations and laboratory markers of patients with potentially pathogenic novel FAS and CASP10 variants are listed in Table 2. FASLG and CASP8 pathogenic mutations were not identified in our definite or suspected patient cohort.
In our ALPS-U patient group, the targeted immunodeficiency and gastrointestinal enrichment NGS panel for immunodeficiency genes identified pathogenic, likely pathogenic, or unknown significant variants in 6 definite and 4 suspected ALPS patients (Table 3). The following genes were affected in our patient cohort: inhibitor of nuclear factor κ B kinase regulatory subunit γ (IKBKG), ORAI calcium release-activated calcium modulator 1 (ORAI1), myosin VB (MYO5B), perforin 1 (PRF1), recombination activating 1 (RAG1), signal transducer and activator of transcription 3 (STAT3), and TNF receptor superfamily member 13B (TNFRSF13B). Lymphoproliferation and elevated DNTs were observed in all cases with available data.
Comparison of ALPS-FAS with ALPS-U
In the combined suspected and definite ALPS groups, ALPS-FAS patients (n = 13 of 17) had significantly higher DNT levels (median, 7.5%; range, 4.5% to 23%) compared with ALPS-U patients (32 of 35; median, 2.7%; range, 1.8% to 11%; P < .0001; Figure 3A). The in vitro apoptosis functional test was more impaired in ALPS-FAS patients (n = 14 of 17; median, 1.6; range: 0.4-3.5) than in ALPS-U patients (n = 31 of 35; median, 3.1; range, 1.3-18.1; P < .0001; Figure 3B). Although sFASL proved to be a highly predictive biomarker for ALPS-FAS (n = 16 of 17; median, >1000 pg/mL; range, 128.9->1000 pg/mL), in ALPS-U, a vast majority of patients showed normal or moderately elevated biomarker levels (n = 29 of 35; median, 152 pg/mL; range, 23.5-486 pg/mL; P < .0001; Figure 3C).
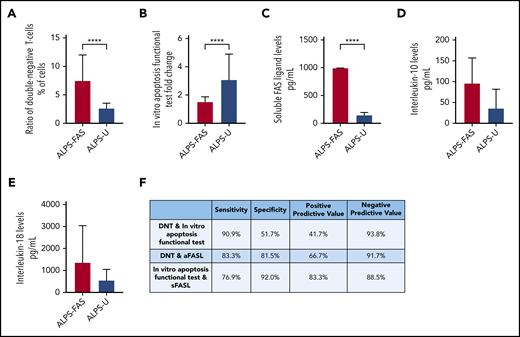
Changes of laboratory parameters in ALPS-FAS and ALPS-U. All suspected and definite ALPS patients were categorized by genetic results as ALPS-FAS or ALPS-U. (A) DNTs. (B) In vitro apoptosis functional test. (C) sFASL. (D) IL-10 (P = .3070). (E) IL-18 (P = .0514). (F) Evaluation of combinations of laboratory parameters to differentiate ALPS-FAS and ALPS-U. All data are presented as median ± interquartile range. ****P < .0001.
Changes of laboratory parameters in ALPS-FAS and ALPS-U. All suspected and definite ALPS patients were categorized by genetic results as ALPS-FAS or ALPS-U. (A) DNTs. (B) In vitro apoptosis functional test. (C) sFASL. (D) IL-10 (P = .3070). (E) IL-18 (P = .0514). (F) Evaluation of combinations of laboratory parameters to differentiate ALPS-FAS and ALPS-U. All data are presented as median ± interquartile range. ****P < .0001.
No significant difference was observed regarding IL-10 (ALPS-FAS: n = 4 of 17; median, 96.8 pg/mL; range, 19.9-169.2 pg/mL vs ALPS-U: n = 15 of 35; median, 36.8 pg/mL; range, 19-167.3 pg/mL; P = .3070), whereas a tendency toward increased IL-18 was found in ALPS-FAS (n = 9 of 17; median, 1380 pg/mL; range, 265-3255 pg/mL) compared with ALPS-U (n = 13 of 35; 570 pg/mL; range, 157-3180 pg/mL; P = .0514; Figure 3D-E).
Examining the marker combinations, the DNT and in vitro apoptosis functional test combination showed the highest sensitivity (90.9%) to differentiate between ALPS-FAS and ALPS-U, with a negative predictive value of 93.8%. In contrast, the in vitro apoptosis functional test and sFASL combination showed the highest specificity, at 92%, with the highest positive predictive value (PPV), at 83.3%, for ALPS-FAS. All 3 combinations showed a significantly higher frequency of abnormal results in the ALPS-FAS group (P = .01465 for DNT and in vitro apoptosis functional test; P = .0002 for DNT and sFASL; P < .0001 for sFASL and in vitro functional apoptosis test; Figure 3F).
Discussion
ALPS is a clinically and genetically heterogeneous disease with nonspecific signs and symptoms. After its description in 1992, several diagnostic criteria were developed to identify the characteristics of this rare disease. All diagnostic protocols from 2000 to date include similar clinical features and biomarkers with different combinations and priorities.2,3,35,36 The latest working definitions for clinical diagnosis of ALPS published in 2019 by European Society for Immunodeficiencies permit a wide range of biomarker combinations and do not mention strictly required criteria, such as DNT elevation or lymphoproliferation.36 Therefore, this diagnostic protocol allows the potential identification of patients with presymptomatic or mild disease or even of those patients who previously received immunosuppressive treatment. DNT, in vitro lymphocyte apoptosis functional test, sFASL, and IL-10 could be in the normal or moderately abnormal range during or after immunosuppressive medications.37,38 Therefore, we extended the diagnostic protocol with the category of suspected ALPS (2 major and 1 minor criterion observed) to include all patients whose manifestation resembled ALPS, even if their biomarkers were moderately abnormal or unavailable. The limitations of our study are that clinical outcome data were collected and biomarkers were analyzed at the time of patient referral; therefore, incidence of splenectomy and lymphoma development over time could not be assessed, and the possibility of previous or ongoing immunosuppressive medication could not be ruled out.
Our findings support the generally accepted diagnostic protocols2,3,36 that lymphoproliferation (with/without splenomegaly and/or lymphadenopathy) is the most important clinical manifestation of ALPS; it was present in 97.1% of our definite ALPS patients, a rate similar to other previous publications.5 Furthermore, all definite and suspected ALPS patients had elevated DNTs, when the test was performed.2,3,36 However, 51.5% of patients with an unlikely ALPS diagnosis also showed abnormal DNT levels, and therefore, the specificity (50%) and PPV of DNTs (43%) proved to be relatively low, as previously published.39 Several types of immunodeficiencies, such as X-linked immunodeficiency with magnesium defect, Epstein-Barr virus infection, and neoplasia,40 autoimmune disorders, such as juvenile systemic lupus erythematosus, mixed connective tissue disease, and severe infections,41,42 can also cause elevated DNTs.43 By contrast, the in vitro apoptosis functional test showed abnormalities in 78.8% of the definite ALPS patients, whereas only 13.8% of the unlikely ALPS population had abnormal apoptosis function. Therefore, this biomarker showed high specificity (92.2%), with high PPV (78.8%). According to our findings, the sensitivity of these biomarkers in combination is 79.3%, and the specificity is 93.7%. The presence of abnormal levels of these 2 markers could support the presence of ALPS, and a diagnosis of ALPS could be ruled out if both biomarkers are within the normal range. Although sFASL was significantly elevated in all ALPS populations, its combinations (DNT and sFASL, in vitro apoptosis functional test and sFASL) presented similar specificity but reduced sensitivity compared with the combination of DNT and in vitro apoptosis functional test. IL-18, IL-10, IgG, and vitamin B12 levels were considered in clinical assessment, but their combinations were not evaluated because of the limited number of cases.
In the suspected ALPS group, in addition to elevated DNTs, a vast majority of biomarkers were moderately abnormal, and a significant difference was seen only in in vitro apoptosis function, with a tendency toward significance in sFASL, compared with the definite ALPS population (Figure 2C-D). These results could have been caused by previously commenced immunosuppressive medication or the early development of disease.
In our study investigating a large number of patients with FAS gene mutations, 17 cases from 10 independent families were identified. Pathogenic or likely pathogenic FAS gene mutations could either cause haploinsufficiency (nonsense and frameshift variants) or disturb the interaction of death receptor FAS and the adaptor protein FADD oligomers. All pathogenic missense mutations, including the novel variants in our study, affected the death domain of the FAS protein (c.742-c.826), responsible for a strong dominant negative effect.44 The only nonsynonymous missense mutation in our cohort occurring outside of the FAS domain (c.136A>G) occurred in an unlikely ALPS patient, reducing the possibility of pathogenicity of the variant (although Varsome category is VUS). Interestingly, early STOP codon–producing nonsense FAS mutations produced discrepant symptoms in our ALPS patient cohort. For example, patient 39 had clinical symptoms and laboratory results supporting an ALPS diagnosis, except the apoptosis functional assay. However, patients 107 and 49, who also had nonsense FAS mutations, had impaired apoptosis function. The most probable explanation for this might be that in patient 39, the mutation affected the extracellular region of FAS, causing haploinsufficiency, whereas in patients 107 and 49, the mutations affected the intracellular region, causing a dominant negative effect. It has been previously reported that the intracellular mutations of the FAS receptor have a higher penetrance than extracellular mutations.4,30 In our study, PB samples were used for genetic screening; therefore, somatic mutations were not analyzed in cellular subsets. The coverage of NGS was optimized for germline mutation detection. In the future, it would be desirable to perform FAS gene mutation sequencing in sorted DNT fractions to differentiate between ALPS with somatic FAS mutation and ALPS-U.
In our cohort, 10 of 37 patients categorized as having ALPS-U had variants in genes other than FAS or CASP10. A vast majority of genes identified in our ALPS-U cohort have been previously described in connection with immunodysregulation. Of the identified genes in our ALPS-U cohort, IKBKG, ORAI1, MYO5B, PRF1, and RAG1 gene products do not interact with the Fas-mediated apoptotic pathway according to currently available data. IKBKG and ORAI1 genes play a role in the development of ectodermal dysplasia with immunodeficiency. Interestingly, elevated DNTs were noted in a patient with IKBKG mutation; however, apoptosis functional impairment had not been described in such patients previously.45 The MYO5B gene is related to microvillus inclusion disease characterized by gastrointestinal and neurological symptoms, but it is not related to lymphoproliferation. PRF1 gene mutations are responsible for familial hemophagocytic lymphohistiocytosis, a syndrome sharing common clinical signs with ALPS, such as splenomegaly and cytopenia. Of note, PRF1 c.272C>T and c.755A>G found in our ALPS-U patients are both described as fairly common variants in the healthy population, with an MAF of 2.9% and 0.5%, respectively. The RAG1 likely pathogenic variant (c.2290C>T) is associated with Omenn’s syndrome, an autosomal recessively inherited form of severe combined immunodeficiency. Compound heterozygous RAG1 mutations have also been reported in Evans syndrome.46 Interestingly, our patient was homozygous for the RAG1 variant. The pathogenic gain-of-function mutation in STAT3 (c.2144C>T), identified in 2 unrelated patients in our cohort, leads to lymphoproliferation, autoimmunity, and recurrent infections47 ; the same mutation was independently described in Evans syndrome.46 TNFRSF13B c.310T>C, a common functional variant influencing receptor ligand binding and signaling, occurs in heterozygous forms in 2% to 5% of cases of common variable immunodeficiency and in 0.5% to 1% of healthy individuals.48 As described by Teachey et al,3 common variable immunodeficiency patients can present with ALPS-like features.
All patients with ALPS-FAS showed significantly abnormal levels of a majority of recommended laboratory markers, such as DNTs, apoptosis functional test, and sFASL, similar to previous studies.37,39,49 However, patients belonging to the ALPS-U group showed mostly normal apoptosis function and levels of sFASL. This supports the previous finding that ALPS-U patients do not have a detectable abnormal laboratory marker, which would support the presence of pathogenicity in the extrinsic apoptotic pathway. In addition, the combination of higher in vitro apoptosis functional test score and sFASL level could presumptively predict the ALPS-FAS diagnosis, because the specificity was 92%, and the PPV was 83.3% (Figure 3F).
Pathogenicity predictions applied by the ACMG criteria were rather contradictory in the case of CASP10 missense variants. The occurrence of the variants in the general population with a considerable MAF of 0.04% to 4% and the inconsistent but mostly neutral/tolerated in silico functional predictions resulted in likely benign or benign ACMG classification. Supporting this prediction, CASP10 c.1216A>T (MAF, 4% in the Danish population33 ) cooccurred with another missense CASP10 variant (c.1216A>T) in our cohort. CASP10 c.1216A>T was considered as a VUS/likely pathogenic variant in our patient cohort, because in vitro functional studies have proven defective apoptosis in ALPS patients.28 The CASP10 c295A>G variant has not been previously described in the literature. Biomarker data on ALPS-CASP10 patients are scarce; interestingly, none of our patients showed defective in vitro apoptosis functional assay and elevated sFASL. However, this tendency was not confirmed statistically because of the small number of patients. Patients with CASP10 mutations did not show any abnormalities in in vitro apoptosis function. Therefore, the usefulness of the FAS-mediated apoptosis assay and the measurement of sFASL level could be questioned in the ALPS-CASP10 group and would need further investigation. In line with previous observations, the lack of pathogenic FASLG and CASP8 variants in our patient cohort further supports the rare occurrence of genetic defects in these genes.2,10,18
Our findings illustrate that both major and minor biomarkers defined in the new diagnostic protocol of 2010 are helpful in ALPS diagnosis. However, not all of them show significant abnormalities in all types of ALPS. Those patients who do not present with lymphadenopathy and/or splenomegaly and do not have increased levels of DNTs are unlikely to have ALPS. Because measurement of DNT levels is readily available, these should be assayed as the first investigation in patients suspected of having ALPS. If normal, except in cases of high clinical suspicion, further investigation is not required. Several other nonimmunological parameters, such as vitamin B12 or high-density lipoprotein, have been investigated as useful biomarkers.39,49 The abnormal synthesis and release of haptocorrin, 1 of the vitamin B12 transport proteins, from ALPS lymphocytes are responsible for the high vitamin B12 in ALPS,50 whereas elevated IL-10 was reported as the direct cause of serum lipoprotein alterations.51 A limitation of our study, these nonspecific but easily available biomarkers (eg, high-density lipoprotein) were not recorded in all cases; therefore, analyses could not be conducted here.
Our findings support that an elevated DNT level is an essential major criterion for ALPS (but not a required criterion, if the measurement of DNTs is not available). The use of biomarker combinations (DNTs, in vitro apoptosis functional test, and sFASL) could accelerate the confirmation or exclusion of an ALPS diagnosis, which is especially useful when molecular analysis is also not available. These reliable blood biomarkers could act as substitutes for molecular analysis by a gene panel that includes FAS and CASP10, decreasing the cost of diagnosis. Genetic sequencing could be reserved for those patients who present with uncharacteristic combinations of these biomarkers and have high clinical suspicion of ALPS. This diagnostic consideration could facilitate and simplify the rapid diagnosis and treatment of ALPS and decrease the cost of the diagnostic process. The results from this report should help guide targeted combinations of biomarkers, resulting in rapid diagnosis and the optimal use of resources.
Contact the corresponding author for original data.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors acknowledge the staff of the Great Ormond Street Hospital Immunology Laboratory and the North East Thames Regional Genetics Service for helping to analyze these samples.
This research was supported by the Wellcome Trust and by a National Institute for Health Research Great Ormond Street Hospital UCL Biomedical Research Centre award. E.M. and G.K. were supported by the European Union and the Hungarian government (EFOP-3.6.3-VEKOP-16-2017-00009).
Authorship
Contribution: E.M. and N.R. analyzed the data and wrote the paper; E.M., N.R., and A.Z. collected clinical and laboratory results; F.H., M.H., D.L., and K.C.G. helped to establish the assays and translate into routine service; G.K. and H.A. performed the statistical analysis and assisted with the evaluation of molecular analysis; A.J.T., F.H., M.B., S.O.B., and K.C.G. supervised the work and gained the funding under which this work was performed; and all authors reviewed and edited the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Kimberly C. Gilmour, Great Ormond Street Hospital for Children, NHS Foundation Trust, Great Ormond St, London WC1N 3JH, United Kingdom; e-mail: kimberly.gilmour@gosh.nhs.uk.


