Abstract
Using whole-exome sequencing to examine the genetic causes of immune deficiency in 235 common variable immunodeficiency (CVID) patients seen in the United States (Mount Sinai, New York), 128 patients from Sweden, and 208 from Iran revealed 68 known disease-causing genes underlying this heterogeneous immune defect. The patients at the time of study ranged from 4 to 90 years of age. Overall, 31%, 36%, and 54% of the patients in the US, Swedish, or Iranian cohorts had mutations. The multiplicity of genes identified in the 571 subjects reflects the complex requirements of B-cell antigen signaling, activation, survival, migration, maturation, and maintenance of antibody-secreting memory B-cell populations to the plasma cell stage. For the US and Swedish cohorts, CVID subjects with noninfectious complications, lymphoid infiltrations, inflamatory conditions, or autoimmunity were somewhat more likely to have an identifiable gene, but in both cohorts, numerous subjects with these medical conditions had no potential gene that could be assigned. Specific clinical patterns of illnesses were also not linked to any given gene defect as there was considerable overlap in clinical presentations. These observations led to a new perspective on the complexity of the immunologic phenotype found in CVID syndrome.
Common variable immunodeficiency (CVID) can best be described as a collection of hypogammaglobulinemia syndromes, first segregated from other immune-deficiency syndromes in 1971 by a panel of experts convened by the World Health Organization in a consensus statement. In the resulting report, the syndrome of patients with “late-onset” immunoglobulin failure was called variable immune deficiency (common, largely unclassified),1 later condensed to “common variable immunodeficiency” (CVI and then later, CVID) to separate this syndrome from other forms of genetic immune defects such as X-linked agammaglobulinemia, Wiskott-Aldrich syndrome, hyper immunoglobulin M (IgM) syndromes, and other defects with syndromic features or obvious Mendelian inheritance. In contrast to younger patients with agammaglobulinemia or hypogammaglobulinemia, these somewhat older subjects were often diagnosed with “acquired hypogammaglobulinemia.” The term acquired, as due to an event after birth, was used simply because of the later onset, but investigators were puzzled over the intriguing question of why the B cells of CVID subjects might lose function over time. However, very soon after the first description, Brem and Morton wrote that they found it difficult to envision any acquired process that would selectively “assault” all globulin production, without affecting other related proteins or functions; on this basis, they suggested that single-gene defects would be more likely,2 which was a prescient view, as the last decade has finally shown.
Although definitions have varied, the diagnosis of CVID is commonly applied to male or female patients with low levels of serum IgG, IgA, and/or IgM, with absent or defective antibody production to both protein and carbohydrate antigens.3-6 The majority of newly diagnosed patients are adults between 20 and 40 years of age.5,7,8 However, many patients have had a clinical history suggestive of an immunodeficiency for some years before the diagnosis of CVID is actually made. In the European Society for Immunodeficiency (ESID) data collection, the mean diagnostic delay was 7.46 years with a range of 0 to 61 years.7 Children younger than 4 years of age are not generally given this diagnosis, as immaturity or other genetic causes are more likely in this young age group. However, if the immune defect persists and no other causes are found, the CVID term can be used. IgG deficiency is also separable from CVID as the immune and clinical phenotypes differ significantly.9 In addition, patients with quite reduced numbers of CD4+ T cells (<200/µL), especially naive T cells, are more likely to have a genetically defined combined immune defect and are often distinguished in case studies.6,10 Overall, CVID is a diagnosis of exclusion, in that other causes of hypogammaglobulinemia, whether primary or secondary, must be excluded; these include bone marrow infiltrative disease, medications, also immune globulin losses (for example in the gut or urine), as well as all other genetic defects.
A curious feature about CVID syndrome is that patients can be divided into 2 major groups based on clinical presentations. The first group has infections only, whereas the second has a variety of noninfectious autoimmune, inflammatory, and/or lymphoproliferative manifestations, also associated with systemic immune activation.8,11,12 In some cases, patients in the second group may have inflammatory features as the initial presentation and their only symptom, with no obvious susceptibility to infectious diseases. Large studies have shown that subjects with the inflammatory/autoimmune features have increased morbidity and mortality, and therefore a number of studies have probed reasons for the striking heterogeneity of these patient groups.8,11,13
Approaching the question of how to differentiate these groups of patients, whole-exome (WES) and whole-genome analyses have now revealed an increasing number of monogenic causes of the CVID phenotype, as originally suggested. The first causes identified were autosomal-recessive (AR) disorders, often in subjects born to consanguineous parents. It was also recognized that biallelic mutations in genes such as adenosine deaminase (ADA), recombination activating 1 and 2 (RAG1 and RAG2), as well as in Artemis, more commonly underlying severe combined immune deficiency, could also produce milder CVID-like defects. More recently, an increasing number of autosomal-dominant (AD) immune defects that lead to hypogammaglobulinemia as a prominent immunologic outcome have been discovered. The genes now identified in the CVID phenotype are diverse, and in some cases have been associated with other syndromes and disease states, but not as defects leading to hypogammaglobulinemia as the presenting feature. However, the identified genes reflect the complex requirements of B-cell antigen signaling, activation, migration, long-term survival, and maturation of antibody-secreting memory B cells to the plasma cell stage. These observations have led to a new perspective on the complexity of the immunologic phenotypes that lead to CVID syndrome.
Here, we describe the results of genetic analysis of a large and heterogeneous CVID patient population, the majority of whom are from the United States (Mount Sinai, New York) and 2 other patient populations: 1 from Sweden and 1 from Iran. These analyses demonstrate the incidence and range of genes that lead to the CVID phenotype, and the differences between these patient populations. From the standpoint of the practicing physician, an important question for genetic analyses is whether the clinical phenotype of patients suggests the likelihood of identifying a causative mutation.
Methods
Patient selection
Subjects were diagnosed with CVID using established criteria, including serum IgG and IgA and/or IgM deficiency with proven loss of antibody production.3,14 For comparisons of different patient populations, subjects were recruited from 3 patient populations: Mount Sinai Medical Center in the United States (New York), the Karolinska Institute in Sweden (Stockholm), and Tehran University of Medical Sciences in Iran (Tehran). The subjects selected for WES were also classified into 1 of 2 groups of patients: those who had infections as their only manifestation of immune deficiency, and subjects with inflammatory/autoimmune complications as previously described.8,11 In a minority of cases for the US and Swedish cohorts, other members of a family were affected, and these are distinguished in the results. In contrast to previous reports, which have described antibody-deficient patients with AR variants,15 no subjects in the US or Swedish cohorts presented here had a known background of consanguinity. When available, samples from parents and siblings of CVID patients were submitted for WES and/or Sanger sequencing to study familial segregation. Ethical permissions were obtained from the ethics committees of the participating centers. Informed consent was obtained from all individuals and/or their legal guardians.
WES
Genomic DNA was extracted from peripheral blood mononuclear cells and sheared with a Covaris S2 Ultrasonicator. An adaptor-ligated library was prepared with the Paired-End Sample Prep kit V1 (Illumina). Exome capture was performed with the SureSelect Human All Exon kit (Agilent Technologies). Massively parallel sequencing was performed on a HiSeq 2500 (Illumina), which generates 100 base reads. Sequences were aligned for variant calling and annotation with the human genome reference sequence (hg19 build) using the BWA aligner.16 Downstream processing was performed with the Genome analysis toolkit (GATK),17 SAMtools,18 and Picard Tools (http://broadinstitute.github.io/picard/). A GATK UnifiedGenotyper and a GATK IndelGenotyperV2 were used to identify substitution and indel variant calls, respectively. Calls with a read coverage of ≤2x and a Phred-scaled single-nucleotide polymorphism quality of ≤20 were filtered out. All variants were annotated with the GATK Genomic Annotator (Broad Institute). Variants from the Blacklist were excluded.19 Copy-number variant (CNV) analysis was performed using the R software package ExomeDepth (v1.0.7),20 which investigates change in read coverage compared with an expected depth distribution to call CNVs. For each tested individual, the ExomeDepth algorithm builds the most appropriate reference set from the BAM files of a group of samples and ranks the CNV calls by their confidence level.
Targeted gene screening
Patient exomes were filtered for mutations in all known genes associated with a primary immune-deficiency disease, as previously described.14,21 Heterozygous and homozygous mutations were excluded if the allele frequencies in the general population were >0.01% or 1.0%, respectively, in the Exome Aggregation Consortium database (ExAC; Broad Institute) and Genome Aggregation database (gnomAD; Broad Institute). Top likely disease-causing candidates were Sanger sequenced for confirmation. Familial segregation was studied when samples were available. Other candidate mutations were confirmed by examining read alignment in the Integrated Genomics Viewer (IGV; Broad Institute). Variants from the Blacklist were excluded.21 Mutations were subsequently analyzed using computational predictors of mutation severity including combined annotation-dependent depletion22 and were compared with the gene-specific mutation significance cutoff.23 Variants with combined annotation-dependent depletion scores below the gene-specific mutation significance cutoff were excluded. Confirmed variations were also screened through the Human Gene Mutation database24 to identify published disease-associated variations. Therefore, the pathogenicity of all disease-attributable gene variants was evaluated using the updated guideline for interpretation of molecular sequencing by the American College of Medical Genetics and Genomics (ACMG) considering the allele frequency, computational data, immunological/functional data, familial segregation and parental data, and clinical phenotyping.25
Results
Patients
The US cohort of 235 patients included 221 sporadic cases and 14 patients with known familial history of immune deficiency. These included 2 pairs of sisters, 2 pairs of brothers, a father and son, a mother and son, and 2 second cousins. The median age of the US patients was 44 years (range, 5-85 years); 134 were female and 101 were male (Table 1). Of these subjects, 231 were of European descent (which included 7 who were also black) and 4 were Asian. For the 128 subjects in the Swedish cohort, the median age at the time of study was 49.5 years (range, 4-90 years); 63 were male and 65 were female. For the Iranian cohort of 208 subjects, the age was substantially younger, with a median age of 9 years (range, 1-65 years), with 113 males and 95 females.
Mutations identified
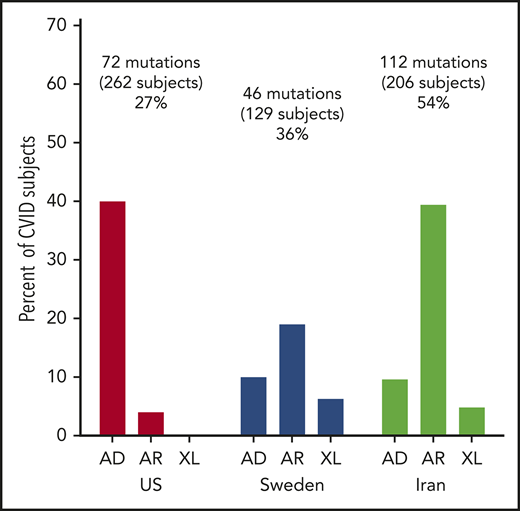
For the US cohort, 74 subjects (31% of the group), of age range 5 to 73 years, with a median age of 35 years, had mutations considered deleterious. The most common variations were in the transmembrane activator and CAML interactor (TACI) gene in 26 subjects, as noted previously in other reports26,27 (Table 2). Fourteen subjects had AD mutations in nuclear factor κ B subunit 1 (NFKB1), as noted in this cohort, and also in other series as underlying AD CVID.21,28,29 More than 1 subject in the US cohort had mutations in other genes previously found in subjects with a CVID phenotype. These included heterozygous mutations in the genes cytotoxic T-lymphocyte associated protein 4 (CTLA4),30,31 IKAROS family zinc finger 1 (IKZF1),32-34 phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit Δ (PIK3CD),35,36 signal transducer and activator of transcription 3 (STAT3),37 phosphoinositide-3-kinase regulatory subunit 1 (PIK3R1),38 transcription factor 3 (TCF3),39 and AR compound heterozygous mutations in lipopolysaccharide-responsive beige-like anchor protein (LRBA).40,41 Four adult subjects with infections, autoimmunity, and mild retardation had mutations in lysine methyltransferase 2 (KMT2D), a gene associated with Kabuki syndrome, a pediatric syndrome of unusual facial features, skeletal abnormalities, mental retardation, and cardiac defects. Two sisters in this cohort with no warts and moderate neutropenia, severe autoimmune thrombocytopenia, and autoimmune hemolytic anemia had frameshift mutations in C-X-C motif chemokine receptor 4 (CXCR4), a gene associated with warts, hypogammaglobulinemia, infections, and myelokathexis (WHIM) syndrome. Eight other subjects had mutations underlying AD, AR, or X-linked (XL) disorders (Table 2).
In the Swedish cohort of 128 subjects, 46 patients (36% of the group), 4 to 90 years old (median age, 49 years), had deleterious mutations. Here, LRBA was the predominant gene identified in 5 subjects (Table 3); 5 patients had heterozygous variations in the TACI gene. More than 1 subject in this cohort had biallelic mutations in the following genes: mismatch repair endonuclease gene MS1 homolog 2 (PMS2; mismatch repair system component), dedicator of cytokinesis 8 (DOCK8), protein unc-93 homolog B1 (UNC93B); compound heterozygous mutations in RAG1/2; or heterozygous mutations in TERF1 interacting nuclear factor 2 (TINF2). X-linked mutations were found in 6 patients, 4 with forkhead box P3 (FOXP3) and 2 with mutations in Bruton tyrosine kinase (BTK). Nineteen other patients had different mutations underlying other AD, AR, or XL disorders (Table 3).
In the Iranian cohort of 208 subjects, mutations leading to the CVID phenotype were identified in 112 subjects (54%), a larger group than in the US or Swedish groups (Table 4). With consanguineous enrichment, the majority of these were, not unexpectedly, biallelic mutations: 15 subjects had biallelic mutations in LRBA; another 8 had mutations in DNA methyltransferase 3 β (DNMT3B; a gene necessary for establishing DNA methylation patterns during development). In another 5, mutations were found in the gene zinc finger and BTB domain containing 24 (ZBTB24; also important for DNA methylation). Mutations in both DNMT3B and ZBTB24 genes are associated with immunodeficiency, centromeric instability, and facial dysmorphism syndrome, a pediatric defect characterized by mild facial dysmorphism, growth retardation, failure to thrive, retardation, recurrent infections, and low serum immunoglobulins.42 Four other subjects had heterozygous mutations in the TACI gene. AR mutations involved in B-cell activation were found in 3 subjects each: CD27 (activation molecule on T and B cells) and PIK3R1, PMS2, RAG1, and Rac family small GTPase 2 (RAC2). As in the Swedish cohort, hemizygous mutations in BTK were found in 5 Iranian patients. Two patients had mutations in 15 additional genes, and 25 other patients had mutations in quite different genes, not identified in others in this group.
Genetics and clinical phenotypes
One of the questions that arises in the care of patients is whether subjects with autoimmune, lymphoproliferative, and/or enteropathic complications have an increased likelihood of being identified with mutations in 1 or more of the genes discussed here. Table 5 outlines the results for these 3 cohorts, dividing subjects according to whether the patient in each group had infections as their only manifestation of immune deficiency, or whether they had had 1 or more of the complications of CVID: polyclonal lymphocytic infiltrations, notable splenomegaly, lymphadenopathy, lymphocytic lung infiltrates (also including granulomatous infiltrates in any location), noninfectious enteropathy, and/or autoimmunity. First, for subjects with infections only, for both the US and Swedish cohorts, approximately one-quarter of these patients had identifiable genes. For those patients with known inflammatory complications, the yield was somewhat higher (35% to 37% of cases). However, for many CVID patients, even those with significant numbers of complications, 65% to 67% of either of these cohorts did not have an identified gene mutation. For the Iranian cohort, more subjects overall had an identifiable genetic cause, and those with or without complications had a similar likelihood of having an identifiable gene. A second question is whether the clinical conditions of patients (infections only, lymphocytic hypertrophy, enteropathy, or autoimmunity) suggest which mutation might be present. Examining the medical records of patients with mutations in the same gene for the more similar US and Swedish cohorts, it is clear that these conditions would not have been useful indicators, as different presentations and different genetic mutations were identified for each gene (Table 6).
Discussion
Whole-exome analyses in 3 different CVID patient groups from 3 different continents revealed 68 known disease-causing genes underlying an immune defect presenting as a CVID syndrome. Although some of the same genes were identified in each group, for the great majority of cases, different genes were identified in each of the cohorts, illustrating the different ethnicities, ages, and degrees of consanguinity of patients seen at these centers. We had expected that the US and Swedish cohorts, with similar median ages and with no history of consanguinity, would be more similar, but aside from variants in the TACI gene, this was not observed, as the most common gene in Sweden was the LRBA gene (followed by several XL genes, PMS2 and DOCK8 mutations). In the US cohort, the second most common gene defect was in the NFKB1 gene, also noted as the most prevalent gene affected in another European cohort.29 However, with the prevalence of NFKB1 variants in the population, it should be noted that not all mutations in the NFKB1 gene have been proven experimentally to be disease-causing. As TACI gene variants commonly found in CVID subjects can be observed in first-degree relatives and healthy individuals with normal immunoglobulin levels,43,44 these may be more disease-modifying than disease-causing, but still, are often associated with lymphoproliferation and autoimmunity.26,43 It is also important to point out that, in addition to these instances, the other genes identified here have also not been experimentally proven to be disease-causing.
Although the first identified genetic etiologies of CVID were AR disorders, as previously summarized,45 increasing numbers of AD defects now constitute the majority of genetic defects identified in CVID patients with nonconsanguineous backgrounds. For the US cohort, the 13 most common defects were heterozygous AD mutations; here, as for other reports cited, affected family members were often asymptomatic. For the Swedish cohort, there were fewer AD disorders, in favor of either XL or AR inheritance.
In each cohort, a few males were identified with known defects in XL genes, including BTK, CD40L, XIAP, FOXP3, SH2D1A, and/or WAS. The other gene mutations identified were variants reported elsewhere in groups of CVID patients; as expected, these include genes known to be important for B-cell activation, including NFKB1, NKFKB2, PIK3CD, PIK3R1, TCF3, CD19, IKBKG, and BLNK, etc. We also point out that each cohort included subjects with gene defects that are associated with other particular syndromes; but these cases did not present with the constellation of syndromic features that would have pointed to these alternative diagnoses. These included mutations in KMT2D, associated with Kabuki syndrome, in a 10-year old and 4 adults with hypogammaglobulinemia. Defects in terminal B-cell differentiation have been described in this syndrome.46 Two hypogammaglobulinemic sisters had mutations in CXCR4, but mild neutropenia, no warts, and recurrent episodes of thrombocytopenia and hemolytic anemic leading to splenectomy. Other examples include variants in genes important for DNA methylation, DNMT3B and ZBTB24, in 1 patient in the Swedish cohort and in 13 patients in the Iranian cohort; as noted in “Mutations identified,” both genes are usually associated with the facial dysmorphism syndrome. For these reasons, although the clinical phenotype may in some cases lead to targeted genetic sequencing of any suspected underlying genes, the mutations in genes identified in our cohorts resulted in either modified, atypical, or alternative clinical and immunologic phenotypes. Overall, these data reflect the fact that the genetic pathways needed for both normal B-cell development, and the long-term maintenance of B-cell memory populations are numerous and complex, demonstrating that mutations in many genes can result in hypogammaglobulinemia and a CVID phenotype. Note, however, that results of the genetic studies can lead reassignment of the patient to the correct immune-defect category, no longer accurately considered to have CVID.
Most genetic studies on primary immune deficiency have concentrated on children. However, a particular point that arises from study of the US and Swedish cohorts is that the age of a subject with a disease-causing variant was not relevant, as monogenic defects were found in CVID subjects of all ages in both the US and Swedish cohorts. For the US cohort, gene defects were found in 74 subjects, of age range 5 to 73 years, with a median age of 35 years. For the Swedish cohort, gene defects were identified in 46 subjects, with an age range of 4 to 90 years, and a median age of 49 years.
The genetic advances in CVID have been remarkable in that although the syndrome is remarkably heterogeneous, single-gene defects do clearly underlie the immune defect of at least one-third or more of patients, depending on the cohort examined. However, it is nonetheless true that, for the majority of patients, genetic defects have not been identified. For some of these, additional novel monogenic causes are likely to be discovered. It is also possible that CVID syndrome results from unrecognized digenic, or oligogenic, causes, which could be dissected by careful consideration of the intersecting immunologic pathways. So far, 1 recognized digenic cause is defects of RAG1/RAG2, which would clearly affect the same pathway; many other cases of compound digenic immune defects are likely. Clearly, for subjects with AD defects, variable penetrance is a reason given for the lack of a clear inheritance pattern. However, even in these cases, we must consider the view that CVID and its clinical manifestations may be produced by the interactions of many genes, possibly with intersecting environmental factors or even epigenetic causes.47,48
The observations reported and discussed here suggest an oligogenic origin for CVID in many patients, where the disease is caused or modulated by other genes, such as, for example, the C-type lectin-like domain family member (CLEC16A)49 or genes in the HLA region.50 Beyond the identification of monogenic causal genes, group clustering studies may prove useful in detecting critical pathways related to the development of the disease, thus contributing to a better understanding of its etiology. In this case, the goal is not proposing the candidate gene(s) in a particular patient and/or doing targeted sequencing in a panel but to take a more nonbiased genome-wide approach in an attempt to detect genes enriched for rare functional variations in the cohort of CVID cases, as compared with healthy controls.
Conclusions
Using WES to examine the genetic causes of immune deficiency in 3 groups of CVID patients from different countries revealed 68 known disease-causing genes underlying this heterogeneous immune defect. Overall, 31%, 36%, and 54% of the patients in the US, Swedish, or Iranian cohorts had such mutations. For the US and Swedish cohorts, CVID subjects with lymphoid infiltrations or inflammatory or autoimmune complications were more likely to have an identifiable gene, but in both cohorts, the majority of subjects with these medical conditions had no potential gene that could currently be assigned.
Acknowledgments
These studies would not have been possible without the genetic, informatic, and computer skills of Patrick Maffucci, Bertrand Boisson, and Mingyan Fang. The authors are particularly grateful to Asghar Aghamohammadi and Jean-Laurent Casanova for their expertise, genetic skills, immunologic expertise, and editing, and for the overall support of this collaboration.
This work was partially supported by the National Institutes of Health, National Institute of Allergy and Infectious Diseases (AI-061093 and AI-08603).
Authorship
Contribution: H.A. performed genetic analyses and defect validation and recruited and cared for patients; L.H. performed genetic studies, designed the study, and edited the manuscript; and C.C.-R. designed the study, performed genetic analyses, recruited and cared for patients, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Charlotte Cunningham-Rundles, Departments of Medicine and Pediatrics, Precision Immunology Institute, The Icahn School of Medicine at Mount Sinai, 1425 Madison Ave, New York, NY 10029; e-mail: charlotte.cunningham-rundles@mssm.edu.