Abstract
Immune thrombocytopenia (ITP) is the most common acquired thrombocytopenia after chemotherapy-induced thrombocytopenia. Existing guidelines describe the management and treatment of most patients who, overall, do well, even if they present with chronic disease, and they are usually not at a high risk for bleeding; however, a small percentage of patients is refractory and difficult to manage. Patients classified as refractory have a diagnosis that is not really ITP or have disease that is difficult to manage. ITP is a diagnosis of exclusion; no specific tests exist to confirm the diagnosis. Response to treatment is the only affirmative confirmation of diagnosis. However, refractory patients do not respond to front-line or other treatments; thus, no confirmation of diagnosis exists. The first section of this review carefully evaluates the diagnostic considerations in patients with refractory ITP. The second section describes combination treatment for refractory cases of ITP. The reported combinations are divided into the era before thrombopoietin (TPO) and rituximab and the current era. Current therapy appears to have increased effectiveness. However, the definition of refractory, if it includes insufficient response to TPO agents, describes a group with more severe and difficult-to-treat disease. The biology of refractory ITP is largely unexplored and includes oligoclonality, lymphocyte pumps, and other possibilities. Newer treatments, especially rapamycin, fostamatinib, FcRn, and BTK inhibitors, may be useful components of future therapy given their mechanisms of action; however, TPO agents, notwithstanding failure as monotherapy, appear to be critical components. In summary, refractory ITP is a complicated entity in which a precise specific diagnosis is as important as the development of effective combination treatments.
Introduction
Immune thrombocytopenia (ITP) is an autoimmune bleeding disorder with thrombocytopenia resulting from increased platelet destruction and inhibition of platelet production.1-4 Most children with ITP have good outcomes with a substantial rate of spontaneous improvement, and those who require intervention or progress to chronic disease usually respond well to treatment. Adults with ITP do not improve as often as children, but they have a higher rate of improvement than generally recognized, perhaps as much as 40% over 1 year and 60% over 3 years.5 Most patients can usually be managed with conventional treatment.1,6 However, small groups of patients exist who are very difficult to manage and do not respond to any treatment (ie, have refractory disease).
Current treatment of ITP is not strictly regimented.7 First-line therapy usually consists of steroids (high-dose dexamethasone or prednisone) or IV immunoglobulin (IVIG), or even a combination of both for certain patients. Second-line treatment primarily includes thrombopoietin receptor agonists (TPO-RAs) and rituximab, with splenectomy deferred until ≥1 y from diagnosis. Additional second-line agents include fostamatinib and immunosuppressive agents (eg, azathioprine, cyclosporine, mycophenolate mofetil, and others). There are no guidelines to specify the order in which second-line agents should be used. The American Society of Hematology guidelines suggest TPO-RAs be used as the first second-line agent in patients with persistent disease. In patients with refractory disease, a number of agents are likely to have been used, including steroids, IVIG, TPO-RAs, rituximab, and/or others, whereas splenectomy will not necessarily have been performed.
Refractory ITP
Defining refractory as “no response to treatment” is subjective.8 We will use the definition of response as outlined by Rodeghiero et al, achieving a platelet count of 30 000/µL and doubling baseline platelet counts.9 Ideally the treatment would be repeated to enhance validity of the lack of response. Failure to respond to splenectomy is included in the definition of “refractory” according to Rodeghiero et al, although this is disputed in children. Currently, there is increasing reluctance to undergo or recommend splenectomy among patients and physicians,10 such that refractory needs to be defined without reference to splenectomy. Furthermore, there is a reluctance to pursue splenectomy when other treatments have been ineffective, based on the not well-documented but widely believed consensus that splenectomy will likely not be effective in such a circumstance.11 Thus, splenectomy may not be performed in otherwise refractory patients. Therefore, we reserve the description of “refractory” for patients whose platelet counts do not respond to ≥2 treatments, there is no single medication to which they respond, and their platelet counts are very low and accompanied by bleeding. These refractory patients have not necessarily undergone splenectomy. Unlike the great majority of patients with ITP, refractory patients do not do well; they respond poorly to a variety of treatments, they develop worsening disease and medication-induced toxicities, they have markedly reduced quality of life, and they have a higher hemorrhagic and infectious morbidity and mortality. The most common reason for medication toxicities in these patients is using steroids at a very high dose or for a very prolonged course.
Very low platelet counts can predispose patients to serious bleeding; however, typical patients with ITP rarely manifest with serious bleeding, even with very low platelet counts.12 A manual count of the platelets in patients with severe thrombocytopenia is typically required, especially with older autoanalyzers, which could be less accurate. Modern autoanalyzers, although not perfect, are more accurate in these cases. Risk factors associated with a high incidence of bleeding are older age, certain comorbidities, need for antiplatelet agents or anticoagulation,13 polypharmacy, and refractory ITP (ie, patients not responsive to many different treatments with very low platelet counts).14 Patients with refractory ITP require vigilant care because of their substantial risk for serious hemorrhage, especially in older age. One hypothesis for the aging effect on the incidence and severity of bleeding is the absence of tonic growth factor nourishment from platelets to aging endothelial cells (eg, VEGF); thus, the endothelium becomes fragile, permitting hemorrhage.13,15,16
These uncommon and very difficult to manage patients are the focus of this review. They are variously described as severe, chronic, refractory, or very-difficult-to-treat patients with ITP. There are 2 parts to the discussion. The first explains the identification and diagnosis of refractory ITP, which is much more complicated than the diagnosis of “common” cases of ITP. The second part describes combination treatments that have been tried in refractory patients. These 2 seemingly disparate topics are united here because they represent the 2 primary considerations for the diagnosis and management of refractory ITP: either the thrombocytopenia is not actually ITP or it is indeed a very-difficult-to-manage refractory ITP.
Diagnosis
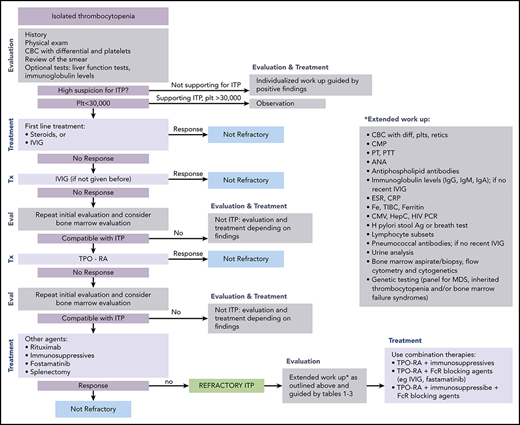
ITP is a diagnosis of exclusion because no specific test defines its presence.17,18 At diagnosis, recommended laboratory testing is a complete blood cell count (CBC) with differential and review of the smear plus/minus immunoglobulin levels, as well as hepatitis C and HIV testing. The general practice of performing only a limited number of tests creates a higher likelihood of an incorrect diagnosis. In a large series of cases seen by experienced hematologists, the 2 leading misdiagnoses were secondary ITP and myelodysplastic syndrome (MDS).19,20 Other misdiagnoses included inherited thrombocytopenia, drug-induced thrombocytopenia, and presentation of bone marrow failure with primarily thrombocytopenia (Tables 1-3).21-24 Figure 1 presents an estimate of primary ITP vs other diagnoses in patients thought to have “refractory ITP.” Response to treatments, especially IVIG, is the only criterion allowing diagnosis of ITP with a high degree of certainty (however, the degree of response required to have a high degree of certainty remains ill defined). In contrast, there is no reliable way to confidently diagnose patients with refractory ITP, because (by definition) the patient does not respond to standard ITP treatment. Figure 2 provides a flowchart of the identification and then the diagnosis of refractory ITP.
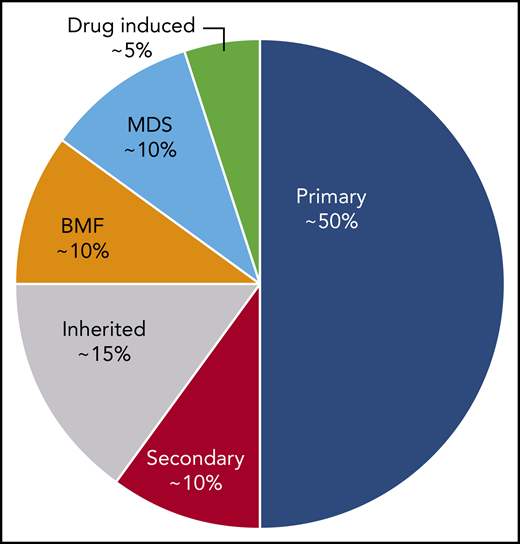
An estimate of the incidence of primary ITP vs other diagnoses in patients defined as having “refractory ITP.” These percentages may vary considerably depending on the clinical setting and geographical location. BMF, bone marrow failure syndromes.
An estimate of the incidence of primary ITP vs other diagnoses in patients defined as having “refractory ITP.” These percentages may vary considerably depending on the clinical setting and geographical location. BMF, bone marrow failure syndromes.
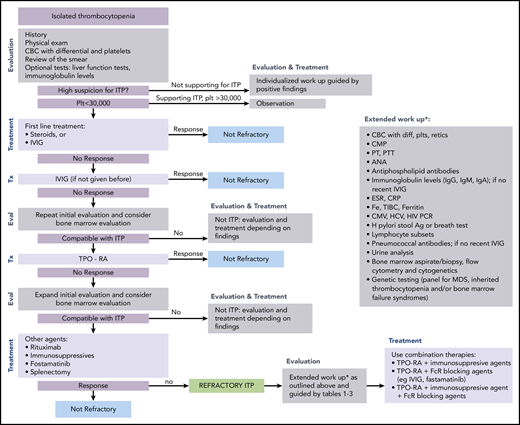
Flowchart for the identification and treatment of patients with refractory ITP. Ag, antigen; ANA, anti-nuclear antibodies; CMP, comprehensive metabolic panel; CRP; C-reactive protein; CMV, cytomegalovirus; ESR, erythrocyte sedimentation rate; Eval, evaluation; HCV, hepatitis C virus; H pylori, Helicobacter pylori; plt/Plt, platelets; PT, prothrombin time; PTT, partial thromboplastin time; TIBC, total iron binding capacity; Tx, treatment.
Flowchart for the identification and treatment of patients with refractory ITP. Ag, antigen; ANA, anti-nuclear antibodies; CMP, comprehensive metabolic panel; CRP; C-reactive protein; CMV, cytomegalovirus; ESR, erythrocyte sedimentation rate; Eval, evaluation; HCV, hepatitis C virus; H pylori, Helicobacter pylori; plt/Plt, platelets; PT, prothrombin time; PTT, partial thromboplastin time; TIBC, total iron binding capacity; Tx, treatment.
Numerous patients with inherited thrombocytopenia have been reported to be initially diagnosed with “ITP” and subsequently received inappropriate and ineffective treatments, including cyclophosphamide and splenectomy. Furthermore, an estimate, based on the incidence of each disease, is that for every 10 cases of apparent ITP, there should be 1 case of inherited thrombocytopenia. This ratio confirms that we substantially underdiagnose inherited thrombocytopenia cases.
Bone marrow failure syndromes may present primarily with thrombocytopenia. However, these patients often provide subtle clues, such as a high mean corpuscular volume in a CBC or a dysmorphic feature, such as hypoplasia of the thenar eminence.
Secondary ITP requires specific testing because patients often do not exhibit overt evidence of their underlying disease. Examples include common variable immune deficiency (CVID) without a history of infections, autoimmune lymphoproliferative syndrome (ALPS) without substantial lymphadenopathy,25 and cytomegalovirus (CMV) infection26 with only mild transaminitis or atypical lymphocytes on smear. These are all “game changers” in that the specific diagnosis dramatically alters management; however, without the specific diagnosis, the ITP would be difficult, if not impossible, to manage. CVID is treated with maintenance IVIG replacement and rituximab if needed.27 ALPS responds to sirolimus (and mycophenolate mofetil),28 and patients with CMV infection worsen with immunosuppression and require direct treatment of CMV.26 Although all of these are very important to identify, altogether they represent <5% of patients with ITP. Other causes of secondary ITP are also important, and the initial estimate by Cines et al (20% of cases of apparent primary ITP are secondary) has been confirmed by 2 studies in France.14,20,29 Drug-induced thrombocytopenia is rarely diagnosed directly by testing.24 Instead, the diagnosis is confirmed when thrombocytopenia resolves after stopping the offending agent. Interestingly, in clinical practice, this change infrequently affects the platelet count. Also, if a patient has been on the same medication for years, it may be essential to other aspects of the patient’s health or it may be difficult to discontinue because of acquired dependence.
When a previously healthy patient presents with isolated thrombocytopenia, one often suspects ITP. As discussed, the rule of thumb is to do as limited an amount of testing as possible if a patient has isolated thrombocytopenia, there are no findings on history or physical examination suggestive of another process, and examination of the peripheral smear reveals blood cells with normal morphology. To exclude all other etiologies by laboratory testing can be an exhaustive task, highly expensive, and hugely unnecessary for the typical ITP patient. However, for difficult cases, as seen in Tables 1 through 3, there is a long list of differential diagnoses to consider, including inherited thrombocytopenia, secondary ITP, and bone marrow failure syndromes. Therefore, one needs to choose which diagnoses to explore first, and this is often not straightforward. Table 4 lists many diagnostic tests that can be used to identify other underlying etiologies.
If several ITP treatments are administered with minimal or no response, it becomes less likely that the patient has ITP (Tables 1-3). Ideally, a work-up is initiated and continued until another diagnosis is made or ITP is confirmed. The following findings are some that may focus the work-up: (1) a history of recurrent infections suggests immunodeficiency, (2) a first-degree relative with low platelet counts suggests an inherited thrombocytopenia, which may also be supported by examination of the peripheral smear, and (3) mild mental retardation, hypocalcemia, and a right-sided aortic arch suggest DiGeorge syndrome. There are many other possible examples and not all are listed in the tables.
It is tempting to perform whole-exome sequencing (WES) or even whole-genome sequencing (WGS). Although this would identify mutations, especially those responsible for bone marrow failure syndromes, inherited thrombocytopenias, and possibly MDSs, it is far from perfect. A 335-patient series of inherited thrombocytopenias identified definite and probable gene findings in less than half of the cases.22 Among bone marrow failure cases, chromosome fragility (diepoxybutane breakage test) can identify Fanconi anemia (FA), and telomere length can identify dyskeratosis congenita (DC); these are often the initial tests done.30
In refractory patients, a poor response to platelet transfusion is not as helpful diagnostically as a good durable response, because many causes of thrombocytopenia can result in a suboptimal response to platelets. Refractory patients should undergo bone marrow examination, including aspirate and biopsy, cytogenetics, and flow cytometry. However, bone marrow examination findings do not allow diagnosis of ITP; they can only be compatible with it. If the bone marrow is normal, WES or whole-genome sequencing is a reasonable next step. Tables 1 through 3 list many entities and their diagnostic modalities. Given the mantra that large platelets mean ITP and not leukemia, myosin heavy chain 9–related disorders (MYH9-RDs), Bernard-Soulier syndrome (BSS), and other macrothrombocytopenias are often misdiagnosed as ITP, especially if they have falsely low platelet counts (ie, if the platelets are too large to be accurately counted).31,32 As already indicated, inherited thrombocytopenias are commonly underdiagnosed. If a TPO-RA is used and responses are only seen to this agent, it may not be helpful diagnostically, because many forms of inherited thrombocytopenia, as well as certain bone marrow failure states, might respond.33,34 Table 3 refers to bone marrow failure states with predominant thrombocytopenia at presentation. Although these are traditionally thought of as pediatric diseases, a number of cases present in adults. The current practice, not to perform routine bone marrow examinations in patients with newly diagnosed suspected ITP, may delay the diagnosis of these diseases, such as FA, acquired amegakaryocytic thrombocytopenia, and telomeropathies (Table 3). Other diagnoses may resemble ITP more closely, including DC (Table 3), chronic lymphocytic leukemia (CLL; if the lymphocyte count is not high) (Table 1), the X-linked thrombocytopenia form (thrombocytopenia only) of Wiskott-Aldrich syndrome35 (Table 2), and asymptomatic HIV (Table 1). MDS may be confused with ITP because it has a similar combination of hypercellular marrow and increased megakaryocytes; signs of dyspoiesis may not be overt, and progression may be required to clarify the diagnosis. Observing a response to IVIG would exclude some of these cases but not secondary ITP; however, a patient who responds to IVIG would not be considered refractory.
Another category is drug-induced thrombocytopenias. Certain medications are known to cause thrombocytopenia.36,37 As mentioned earlier, there is not readily available testing for this entity, and certain cases (ie, quinine in tonic water) can be overlooked.24 Liver disease can cause thrombocytopenia by a number of mechanisms and resemble ITP38 ; however, typically thrombocytopenia is moderate, and patients usually do not present as having refractory ITP. The incidence of silent hepatic disease can vary enormously depending on geography and the type of population served. Splenomegaly (with/without hepatomegaly) may suggest a body computed tomography scan, which may uncover lymphoma or another malignancy.
A work-up needs to be age (and gender) oriented: for example, CLL and MDS are primarily diseases of the elderly and lupus has a ninefold greater incidence in females, with a distinct peak in early adulthood.39-41 CVID can be seen at any age, but it primarily occurs in patients who are 20 to 50 years old. Studies have emphasized that gain-of-function immune defects can manifest as autoimmunity, presenting as refractory ITP at any age.42 High thrombopoietin (TPO) levels might support bone marrow failure syndromes.43 If a work-up has been completed and does not reveal secondary ITP (Table 1), inherited thrombocytopenia (Table 2), or bone marrow failure (Table 3), this does not ensure that the patient has primary ITP. Each category requires thorough investigation with a wide range of testing, including extensive genomic analyses. Furthermore, certain diagnoses do not have specific testing available that will identify all cases. In summary, it is impossible to unequivocally eliminate all possible etiologies of thrombocytopenia.
With these considerations, if an extensive work-up is negative, it remains very difficult to distinguish “world’s worst ITP” from “not ITP at all.” Ideally, a bone marrow examination would exclude MDS and other bone marrow failure conditions; however, this is not infallible and, in certain patients, repeated bone marrow examinations with up-to-date genomic analyses may be required before specific diagnoses can be clarified.
Combination treatment to manage refractory patients with ITP
If a case truly appears to be refractory ITP, the authors’ experiences suggest that, in 50% of cases, it may still be another diagnosis, depending upon the experience of the hematologist and the extent of the work-up. However, if it appears to be ITP and multiple single agents have failed to stably increase the platelet count, combination treatments are the next step. These have been explored in ITP but have not been well reviewed.
Currently, refractory ITP would include lack of response to rituximab and TPO agents. This is not as uncommon as sometimes assumed. The lack of response to these leading second-line agents is what necessitates the use of combination treatment in many patients.
Table 5 lists combination therapies identified for inclusion. The first group of therapies antedates the availability of TPO agents and are no longer used extensively, but they deserve mention. The first combination treatment of which we are aware was cyclophosphamide and prednisone combined with vincristine (CVP), vincristine plus procarbazine (C-MOPP), or etoposide (CEP). The first treated patient was a woman with ITP in 1981 who had relapsed Hodgkin disease and developed refractory ITP. When she was treated with CMOPP for the Hodgkin disease, the ITP improved and, 10 years later, she was still in remission. A further trial of a selection of CMOPP, CEP, or CVP in 8 refractory patients found 4 complete responses (CRs) and 1 partial response (PR). These patients had failed splenectomy and steroids.44 A follow-up letter emphasized that responders remained in remission and included 4 additional patients, 3 with no response and 1 with a PR.45
In 2009, a trial of R-CVP (rituximab, vincristine, cyclophosphamide, and prednisone; a version of R-CHOP without procarbazine) was initiated with disappointing results: of 8 patients treated, only 4 responded, and they were the same ones who had responded previously to rituximab alone (CR or PR).46 Furthermore, time to relapse was approximately the same compared with when patients received rituximab alone. In addition, patients with no response to rituximab did not respond to R-CVP.
In 2007, in the prerituximab era and pre-TPO era, patients who failed to respond to steroids and/or IVIG received induction therapy, followed by combination maintenance therapy. Induction was IVIG, steroids, and IV anti-D and/or vinca alkaloids. The 18 patients needing maintenance therapy received a combination of danazol and azathioprine, with 13 responses.47 More recently, our anecdotal experience in 5 patients found this combination to be ineffective in patients who failed rituximab and/or a TPO agent. This illustrates that the current functional definition of “refractory” has shifted to patients with ITP that is harder to treat in the era of TPO-RA and rituximab availability.
One attempted approach, combining agents that inhibit different T-cell pathways, was administering, at lower-than-maximal dose, azathioprine, cyclosporine, and mycophenolate mofetil in 19 ITP patients with a range of disease severity. The reasoning behind combining these drugs at lower doses was to increase efficacy while reducing toxicity.48 In the pre-TPO era, they demonstrated a 74% response rate without infections; however, long-term follow-up showed that only 2 patients had sustained off-treatment remission.48
Another pre-TPO era approach was triple therapy in newly diagnosed (40%) and “refractory” (chronic) ITP (60%) patients (N = 20), including 5 with secondary ITP (25%). Triple therapy was dexamethasone (4 days at 40 mg/d), low-dose rituximab (4 weekly doses of 100 mg), and low-dose cyclosporine (2.5-3 mg/kg for 28 days) to complete therapy within 1 month. Four of 12 refractory cases responded and maintained their response for ≥7 months; however, follow-up was limited (<24 months), so further duration of responses is unknown.49 With the exception of “triple therapy,” it is not clear whether any of these regimens is active.
In the TPO era, 1 study50 included 37 patients, with a 6-year median duration of ITP, who had failed a median of 10.5 therapies before being categorized as multirefractory. In 14 patients receiving immunosuppressants alone, only 1 achieved CR, and 13 had no response, whereas the combination of immunosuppressants and TPO agents achieved on-treatment responses in 7 of 10 patients (50% CR, 20% PR). This emphasizes the importance of including TPO agents in combination treatments, even if there has not been a response to them as single treatment.51
A trial performed in China explored 2:1 randomization of 4 low-dose rituximab infusions with the recombinant human TPO (300 mg; 3SBio) administered subcutaneously daily for 14 days vs rituximab alone in a total of 105 patients who were refractory to or relapsing on steroid therapy.52 TPO was used to obtain an immediate effect until rituximab achieved a lasting effect. The combination increased the platelet count earlier and reduced bleeding (45% vs 24% in the first 2 months, P = .03); however, there was no difference in sustained response.
Another combination treatment was explored in 18 patients refractory to IVIG and TPO agents alone, using a combination of romiplostim or eltrombopag to increase platelet production, an immunosuppressant (cyclosporine [n = 14] or mycophenolate mofetil [n = 4] at standard doses) to inhibit T-cell effects, and IVIG as needed to inhibit platelet destruction.53 The combinations resulted in very good responses in 72% of these patients with chronic ITP who had failed a median of 6.5 previous treatments. This study emphasized using agents with different mechanisms of action, including a TPO agent, to achieve best effects. There were minimal side effects, and no severe/serious infection was reported; however, the follow-up was limited.
One randomized trial compared danazol plus all-trans retinoic acid (n = 45 patients) with danazol alone (n = 48 patients). At 1-year of follow-up, 63% of patients on all-trans retinoic acid plus danazol showed a sustained response, whereas only 26% of patients receiving danazol monotherapy were relapse free. This population was not very refractory (ie, they had failed steroids but had not undergone splenectomy), and one third of the patients were taking concomitant medications at baseline. This combination needs to be explored in more refractory patients.54
Given the long-term effects of rituximab alone (40%-60% response lasting 1 year with 20%-30% apparent cure, in typical, not refractory, patients with ITP),55,56 the search for the optimal agent(s) to combine with rituximab continues. Several studies explored the addition of dexamethasone to rituximab. Two studies of patients with ITP at diagnosis (or never treated) by Zaja et al57 and Gudbrandsdottir et al58 initially delivered 4 days of high-dose dexamethasone, followed by 4 infusions of standard-dose rituximab. Initial results combining dexamethasone and rituximab demonstrated higher response rates (50% to 70%) compared with dexamethasone alone (20%-35%), but long-term follow-up was not reported. A set of studies in adults59 and children60 combined 3 (4-day) cycles of dexamethasone and standard rituximab with good clinical results. In particular, women of child-bearing age (including female adolescents younger than 18 years old) appeared to have a high and lasting remission rate (>70% out to 6 years), whereas all other groups did not (remission rates were ≤10% past 1-2 years). These results emphasized the good responses in younger (child-bearing age) women and were confirmed in another study by Marangon et al.61 However, women with chronic refractory disease did not do as well, so the search for what to combine with rituximab in refractory patients continues.
Biology of refractoriness
Patients with very difficult cases of chronic ITP may lose responsiveness to treatment over time; 1 reason could be evolution to MDS. Another possible mechanism is antigen/epitope spread, generating antiplatelet antibodies directed at new platelet antigens.6 Upregulation of “pumps” that expel treatment molecules from inside cells has been reported in refractory patients, demonstrating another way in which resistance to treatment could develop.62 Treatment with cyclosporine repolarizes the membrane and can reverse the activity of certain lymphocyte pumps.63 If ITP converts from primarily antibody driven to T-cell driven, it may become harder to treat. The study by Chapin et al performed in patients during their follow up visits, elucidated a mechanism of rituximab resistance which might be associated with oligo/monoclonal expansion of Vβ T-cell receptor (VBTCR).59 Among long-term responders, only 1 of 10 had oligo/monoclonal VBTCR expansion, whereas oligo/monoclonal populations were seen in 13 of 26 nonresponders.64 Clonal expansions have been reported in other small series of patients with ITP who are unresponsive to different treatments: splenectomy, rituximab, and TPO agents.65,66 The utility of prospective testing for clonality as a marker of refractoriness, the mechanism of refractoriness in oligo/monoclonal patients, and the appropriate approach to these patients remain to be determined. Other possible mechanisms accounting for rituximab resistance in ITP include expansion of long-lived plasma cells in spleen.67,68 The latter has therapeutic implications (eg, antiplasma cell therapies, such as bortezomib, might be useful).69 Focusing on rituximab, studies have suggested that identifying anti-platelet glycoprotein antibodies pretreatment may predict good responses, consistent with a mechanism of reducing/eliminating anti-platelet antibodies.70 Similar findings exist for fostamatinib.71 The absence of these antibodies might predict failure of treatment if antibody-negative cases are not antiplatelet-antibody mediated. The presence of platelet glycoprotein Ib antibodies as markers of drug resistance (eg, to IVIG and steroids) remains controversial.72,73
Combination treatments have been explored in ITP patients closer to diagnosis (Table 6). They are not considered here, despite promising results, because they were used at or very soon after diagnosis; thus, their efficacy in refractory patients is unknown. These include dexamethasone, eltrombopag, rituximab, and/or alemtuzumab.
Finally, other components of combination therapy may be included in the future. The role of rapamycin in blocking the mTOR pathway has been documented in ALPS-related multiple autoimmune cytopenias.74 Recently approved or encouraging ITP therapies include fostamatinib (an inhibitor of SYK); demethylating agents, such as decitabine75 ; FcRn inhibitors; and BTK inhibitors. These agents have novel mechanisms of action, and their role in single-agent treatment of ITP remains to be determined. How to use them in combination treatment will be an important determination, especially if one espouses the view suggested here that using agents with different mechanisms of action is important for combination treatment (note: combining IVIG and IV anti-D, even though they appear redundant, may work because they interact with different Fc receptors). Furthermore, as described above, IVIG has been used with TPO agents and immunosuppressive agents. The mechanism of IVIG in blocking platelet destruction might be simulated by fostamatinib, which blocks FcR signaling. Finally, anecdotal evidence suggests that there may be an enhanced effect by adding romiplostim and eltrombopag to a combination regimen, based on their different binding sites on the TPO receptor, different effects on megakaryocytes and their precursors, and the observation that 50% of patients not responding to 1 drug may respond to the other.76,77 Figure 3 illustrates the different mechanisms of action of medications used for ITP.
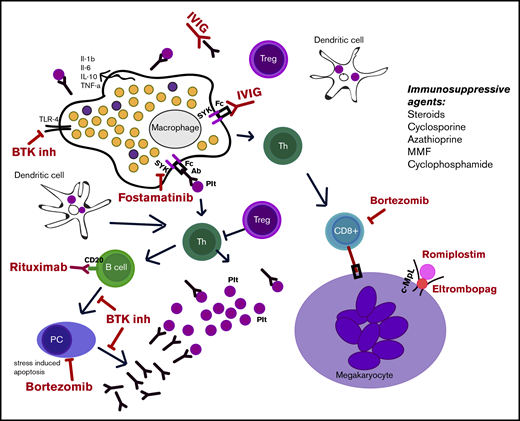
Illustration of the different mechanisms of action of ITP medications. Immunosuppressive agents are also listed. Ab, antibody; BTK inh, BTK inhibitor; c-MpL, thrombopoietin receptor; IL, interleukin; MMF, mycophenolate mofetil; PC, plasma cell; Plt, platelets; Th, helper T cell; TLR-4, Toll-like receptor-4; TNF-α, tumor necrosis factor-α; Treg, regulatory T cell.
Illustration of the different mechanisms of action of ITP medications. Immunosuppressive agents are also listed. Ab, antibody; BTK inh, BTK inhibitor; c-MpL, thrombopoietin receptor; IL, interleukin; MMF, mycophenolate mofetil; PC, plasma cell; Plt, platelets; Th, helper T cell; TLR-4, Toll-like receptor-4; TNF-α, tumor necrosis factor-α; Treg, regulatory T cell.
In summary, the pathogenesis of ITP is heterogenous. The inability to identify critical pathobiologic differences between patients is a major factor limiting the optimization of diagnosis and treatment, which leaves us in a “trial and error” mode to determine effective therapy. The immune state in a patient may change with time and/or treatment. Thus, diversified and individualized therapeutic methods are needed, which include combination treatments to better treat patients with refractory ITP. For these refractory ITP patients, we believe that combination therapy works better than single-agent therapy. Successful combination therapies appear to include a TPO agent and medication(s) with different mechanisms of action that inhibit platelet destruction. The latter may contribute to increased platelet production by ameliorating immune attack on megakaryocytes. There are many unanswered questions regarding combination therapy, including which agents to use and at what dose, how long to give them, and to which patients to give them. Nonetheless, compared with single-agent therapy, combination therapies are more effective in patients with challenging disease. Therapies may have tolerable toxicities, potentially as a result of the ability to use them at slightly lower effective doses. Based on the studies described, we propose the following tenets. First, refractory ITP is even harder to treat in patients unresponsive to TPO agents and rituximab than it was before the advent of these treatments. Second, when choosing agents to combine, select agents with different mechanisms of effect and different primary toxicities (despite the latter not being discussed here); agents active in different parts of the same pathway may have additive or synergistic effects. TPO-RAs appear to be uniquely useful. Third, if a treatment is not effective, instead of stopping it and starting another treatment, it may be better to add the new treatment to the one already being given (ie, initiate combination therapy, despite the lack of effect of the initial treatment). Fourth, based on small studies, oligoclonal/monoclonal T-cell populations may be important biomarkers, indicating a higher likelihood of refractory disease. Finally, patients with the most difficult to treat disease are reasonably likely not to have ITP. MDS and inherited thrombocytopenias would be the most likely “misdiagnoses,”19 but many other possibilities exist; multiple examples have been included. We believe that the identification of a specific cause of thrombocytopenia often results in a specific treatment approach. Ideally, for all cases of ITP, this will be the path forward in the future. Ideally, identification of the pathophysiology in each patient would precede initiation of treatment. Furthermore, because chronic ITP sometimes involves more than just accelerated platelet destruction, ≥2 agents may be required for combination therapy to provide optimal effective management.
Acknowledgment
O.M. acknowledges support from National Institutes of Health, National Cancer Institute grant P30 CA008748.
Authorship
Contribution: All authors contributed to the writing and critical review of this manuscript and agreed on its submission.
Conflict-of-interest disclosure: M.H. has received research support from 3SBio and is a member of the Advisory Board for Novartis and 3SBio. J.B.B. is a member of the Advisory Board for Dova Pharmaceuticals, Amgen, Novartis, Rigel Pharmaceuticals. Inc., UCB, Inc., and Momenta Pharmaceuticals, Inc. O.M. declares no competing financial interests.
Correspondence: Oriana Miltiadous, Memorial Sloan Kettering Institute, Zuckerman Research Center, Z-1419, 408 East 69th St, New York, NY 10021, e-mail: miltiado@mskcc.org.