

Key Points
Few children developed recurrent VTE using dabigatran pediatric formulations as secondary VTE prophylaxis.
Few children experienced major or clinically relevant nonmajor bleeding events when receiving secondary VTE prophylaxis with dabigatran.

Abstract
This open-label, single-arm, prospective cohort trial is the first phase 3 safety study to describe outcomes in children treated with dabigatran etexilate for secondary venous thromboembolism (VTE) prevention. Eligible children aged 12 to <18 years (age stratum 1), 2 to <12 years (stratum 2), and >3 months to <2 years (stratum 3) had an objectively confirmed diagnosis of VTE treated with standard of care (SOC) for ≥3 months, or had completed dabigatran or SOC treatment in the DIVERSITY trial (NCT01895777) and had an unresolved clinical thrombosis risk factor requiring further anticoagulation. Children received dabigatran for up to 12 months, or less if the identified VTE clinical risk factor resolved. Primary end points included VTE recurrence, bleeding events, and mortality at 6 and 12 months. Overall, 203 children received dabigatran, with median exposure being 36.3 weeks (range, 0-57 weeks); 171 of 203 (84.2%) and 32 of 203 (15.8%) took capsules and pellets, respectively. Overall, 2 of 203 children (1.0%) experienced on-treatment VTE recurrence, and 3 of 203 (1.5%) experienced major bleeding events, with 2 (1.0%) reporting clinically relevant nonmajor bleeding events, and 37 (18.2%) minor bleeding events. There were no on-treatment deaths. On-treatment postthrombotic syndrome was reported for 2 of 162 children (1.2%) who had deep vein thrombosis or central-line thrombosis as their most recent VTE. Pharmacokinetic/pharmacodynamic relationships of dabigatran were similar to those in adult VTE patients. In summary, dabigatran showed a favorable safety profile for secondary VTE prevention in children aged from >3 months to <18 years with persistent VTE risk factor(s). This trial was registered at www.clinicaltrials.gov as #NCT02197416.
Introduction
Venous thromboembolism (VTE) in children is associated with considerable morbidity and mortality.1-4 Preventing secondary VTE in children poses a challenge for clinicians, due to the evolving maturation of a child’s hemostatic system with age, which affects not only the risk of recurrent VTE but also the pharmacokinetics and responses to anticoagulants and antiplatelet therapies.5,6 Risk factors for recurrent VTE, the presence of comorbidities, failure to monitor VTE adequately to inform treatment decisions, and limited vascular access (which may impact treatment choice) contribute to treatment complexity.5,6 Risk factors that have been reported to be associated with recurrent VTE in children include central venous access devices, infection, cancer, congenital heart disease, and thrombophilia.1,7,8
Current standard of care (SOC) for the secondary prevention of VTE in children, including low-molecular-weight heparins (LMWHs) or oral vitamin K antagonists (VKAs), depends primarily upon the cause and risk factors of the first VTE event, with recurrent VTE contributing to the severity and duration of anticoagulation.6 However, current SOC has several limitations depending upon the anticoagulant used. For example, LMWH requires parenteral administration, whereas VKA may result in variable effects and low time in therapeutic range due to frequent food- and drug-drug interactions, requiring the need for regular laboratory monitoring to ensure dosing appropriateness of anticoagulation. Moreover, the rarity of pediatric VTE leads to difficulties in designing and managing clinical trials in this setting.9 The treatment of VTE in children is extrapolated from evidence-based recommendations derived from studies performed in adult populations.6 However, the hemostatic system in infants and children is profoundly different from adults. As such, pediatric safety studies are recommended by both the European Medicines Agency (EMA) and the US Food and Drug Administration (FDA).10,11
Some of the limitations with SOC in children with VTE could be overcome by dabigatran, a direct, oral thrombin inhibitor shown to be effective for the treatment and secondary prevention of VTE in adults.12-14 In addition, previous pediatric phase 2 dabigatran VTE trials have reported similar safety and pharmacokinetic/pharmacodynamic relationships to those seen in adults.15-17 In this open-label, phase 3 trial, we report the first safety data on dabigatran etexilate for the secondary prevention of VTE in children aged <18 years, as well as the appropriateness of an age- and body-weight–adjusted dosing algorithm for dabigatran in this setting.
Materials and methods
Trial design
The trial design has been described in detail previously.18 In brief, this open-label, single-arm, safety prospective cohort, phase 3 clinical trial (NCT02197416) (supplemental Figure 1, available on the Blood Web site) is a part of a Pediatric Investigational Plan agreed upon with the EMA Pediatric Committee, and a postmarketing requirement agreed upon with the US FDA. The main objective was to assess the safety of dabigatran etexilate for secondary prevention of VTE; all study outcomes were considered safety related. The current analysis includes the data set and target enrollment18 that fulfills the requirements of the European Union (EU) Pediatric Investigational Plan agreed with the EMA Pediatric Committee, while recruitment continued in order to fulfill additional US FDA requirements. The trial was conducted in accordance with the Declaration of Helsinki and the principles of Good Clinical Practice, and was approved by all investigational site ethics committees. Written informed consent from parents or legal guardians and pediatric patients (if they were of legal age, or if they reached legal age during the trial) was obtained before participation, according to the International Conference on Harmonisation Good Clinical Practice, and the regulatory and legal requirements of each participating country.
The trial was sponsored by Boehringer Ingelheim. Listings of trial committees and investigators are provided in the supplemental appendix. The steering committee and sponsor representatives developed the protocol, supervised the trial, and oversaw any required protocol amendments. The external independent data monitoring committee (consisting of the same members as for the DIVERSITY trial [NCT01895777]) regularly reviewed safety and efficacy data, advising the sponsor on whether the trial should continue, be modified, or be terminated. An independent blinded adjudication committee evaluated all coprimary end points to confirm or refute outcome events. Boehringer Ingelheim coordinated the trial execution, and oversaw the collection, management, and analysis of trial data.
Trial population
Children aged <18 years (stratified by age: stratum 1, 12 to <18 years; stratum 2, 2 to <12 years; stratum 3, >3 months to <2 years) were eligible if they had an objectively confirmed diagnosis of VTE (eg, by compression ultrasound, computerized tomography, or magnetic resonance imaging scans) treated with SOC for ≥3 months, or if they had completed dabigatran or SOC treatment in the DIVERSITY trial and had an unresolved clinical thrombosis risk factor requiring further anticoagulation. Complete inclusion and exclusion criteria are provided in supplemental Table 1.
Treatment
Patients were treated with open-label dabigatran for up to 12 months, or less if the identified clinical risk factor for VTE resolved. An age- and weight-adjusted nomogram according to Hayton,19 derived from estimated renal function, was used to dose dabigatran and achieve comparable exposure to adult populations treated with dabigatran.18 Only 1 dabigatran dose modification (up- or down-titration) according to the nomogram was allowed. Different formulations of dabigatran were administered depending upon the age of the child; capsules were given to those aged 8 to <18 years, pellets to those aged <8 years (or those aged 8 to <12 years unable to swallow capsules); oral solution was offered for those aged from >3 months to <12 months who were unable to swallow pellets (but ultimately no patient in this trial required oral solution). All patients discontinued dabigatran at the end of their trial participation and were switched to SOC if there was a continued need for anticoagulation. After completing trial treatment, patients were followed up for a period of 28 days. Patients who discontinued trial treatment prematurely were to be followed up according to the remaining visit schedule until the end of the trial or at least for 28 days. Therefore, patients who discontinued trial treatment prematurely and had a follow-up period of at least 28 days were not considered as premature trial discontinuations. Patients who completed trial treatment as planned, but did not have a follow-up period of at least 28 days, were considered premature trial discontinuations but not premature treatment discontinuations. Patients who discontinued trial treatment due to resolution of the underlying risk factor were not considered premature treatment discontinuations.
Outcomes
According to the objectives of the study, all outcomes were considered safety related. Primary end points were: recurrence of VTE assessed at 6 and 12 months postenrollment (defined as all recurrent VTE: contiguous progression or noncontiguous new thrombus, including deep vein thrombosis, pulmonary embolism, and paradoxical embolism confirmed by imaging); mortality (overall and thrombotic/thromboembolic event mortality) at 6 and 12 months; major bleeding events (MBEs) at 6 and 12 months (defined as: fatal bleeding; clinically overt bleeding [≥20 g/L decrease in hemoglobin over 24 hours]; retroperitoneal, pulmonary, or bleeding that involves the central nervous system; or bleeding that requires surgical intervention in an operating suite); clinically relevant non-MBEs (CRNMBEs) at 6 and 12 months (defined as: overt bleeding that is not directly attributable to the patient’s underlying medical condition and requires administration of a blood product, or bleeding that requires medical or surgical intervention other than in an operating suite to restore hemostasis); minor bleeding events at 6 and 12 months (defined as any overt or macroscopic evidence of bleeding that does not fulfill the MBE or CRNMBE criteria); and overall mortality and thrombotic/thromboembolism-related mortality.20
Secondary end points included: the occurrence of newly diagnosed or worsening of baseline postthrombotic syndrome (PTS; as per the modified Villalta scale21,22 ) at 6 and 12 months postenrollment; the relationship between plasma dabigatran concentrations (total plasma dabigatran trough levels measured at a central laboratory [Nuvisan GmbH, Neu-Ulm, Germany] by a validated high-performance liquid chromatography–tandem mass spectrometry assay) and pharmacodynamic markers (diluted thrombin time [dTT], activated partial thromboplastin time [aPTT], and ecarin clotting time [ECT] evaluated at a central laboratory [Menal GmbH, Emmendingen, Germany]); the number of patients with dose adjustments; and the acceptability of the administered formulations. The residual effect period for which events were still considered on-treatment following the last intake of trial medication was 3 days.
Statistical analysis
Boehringer Ingelheim was responsible for data collection and statistical analysis, and all authors had access to trial data. Based upon an estimated 5% event rate for the composite of recurrent VTE, major bleeds, and mortality related to thromboembolic event at 12 months, a sample size of 100 patients would provide >99% probability observing at least 1 event. However, if the event rate was 1%, it would still provide >63% probability of observing at least 1 event. Although designed to evaluate 100 patients, the sample size was subsequently increased to 200 patients to meet FDA regulatory authority requests. For the primary end points and the secondary PTS end point at 6 and 12 months postenrollment, time-to-event analyses were summarized as Kaplan-Meier estimates, along with descriptive rates. Subgroup analyses included age strata and sex. For the descriptive analyses, patients with early withdrawal or those who were lost to follow-up were deemed nonevents. Patients were censored from the survival analyses if they withdrew early, were lost to follow-up, or did not have VTE or bleeding episodes. A sensitivity analysis was also conducted including all patients entered in the study and the full study period from entry until the day of the last follow-up visit, or until they were lost to follow-up, death, or consent was withdrawn (“on-treatment plus follow-up”). The pharmacokinetic/pharmacodynamic full analysis set consisted of all patients with ≥1 postbaseline measurement. Pharmacokinetic results were summarized descriptively. Pharmacokinetic/pharmacodynamic relationships were explored using graphical analyses. Safety and adverse events were summarized descriptively.
Results
Participants and follow-up
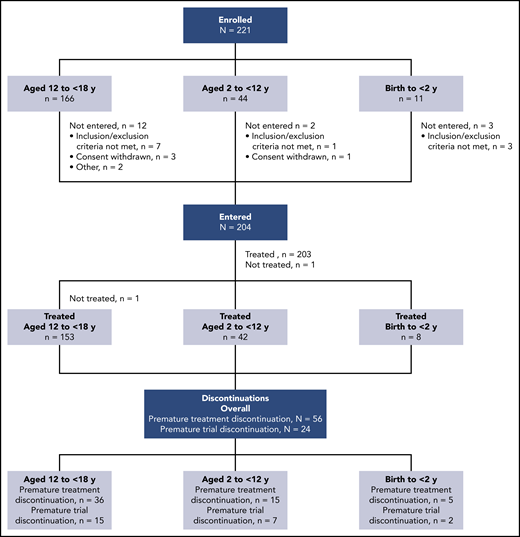
At EU database lock in March 2019, 204 patients from 60 sites in 22 countries had entered the trial, with 1 adolescent not treated (due to inability to take treatment) (Figure 1). The median exposure to dabigatran was 36.3 weeks (range, 0-57 weeks). Risk factors resolved in 32 children during the trial, leading to treatment discontinuation. In 56 of 203 children (27.6%), treatment was discontinued prematurely for the following reasons: target dabigatran concentration not achieved after 1 dose modification (n = 25), noncompliance with protocol (n = 4), VTE recurrence (n = 3; however, 1 of these VTE events was not confirmed to be a VTE recurrence by the adjudication committee and therefore was not included in the analysis), worsening of other preexisting disease (n = 2), other adverse events (n = 4), consent withdrawn regarding dabigatran treatment (n = 2), and other (n = 16). The trial was discontinued prematurely (28-day follow-up period not completed as planned) in 24 of 203 children (11.8%), with reasons being adverse events (n = 3), noncompliance with trial protocol (n = 2), consent withdrawn (n = 5), and other (n = 14). Only 4 patients discontinued both treatment and trial prematurely. All other patients who discontinued the trial prematurely completed the treatment period as planned, but discontinued the trial during the follow-up (posttreatment) period. Baseline demographics are shown in Table 1. Of the 203 treated children, 115 (56.7%) were children receiving chronic anticoagulation who were newly exposed to dabigatran, and 88 (43.3%) were rolled over from the DIVERSITY trial (59 [29.1%] previously treated with dabigatran and 29 [14.3%] with SOC). No children required oral solution; 171 of 203 (84.2%) and 32 of 203 (15.8%) took capsules and pellets, respectively. Overall, 113 of 203 children (55.7%) were male, 101 of 203 (49.8%) were Central European, and 185 of 203 (91.1%) were white. LMWH was the most frequently used prior anticoagulant, prescribed for 152 of 203 children (74.9%). Deep vein thrombosis other than central-line–related thrombosis and cerebral venous thrombosis was the most frequent VTE, reported in 154 of 203 children (75.9%); 35 of 203 children (17.6%) had PTS at baseline.
Baseline patient demographics and characteristics by age strata
| Dabigatran | Total, N = 203 | |||
|---|---|---|---|---|
| 12 to <18 y, n = 153 | 2 to <12 y, n = 42 | 0 to <2 y, n = 8 | ||
| Dabigatran formulation,*n (%) | ||||
| Capsules | 153 (100.0) | 18 (42.9) | 0 | 171 (84.2) |
| Pellets | 0 | 24 (57.1) | 8 (100.0) | 32 (15.8) |
| Age, mean (SD), y | 15.1 (1.6) | 7.0 (3.0) | 0.6 (0.5) | 12.8 (4.5) |
| Male, n (%) | 87 (56.9) | 21 (50.0) | 5 (62.5) | 113 (55.7) |
| Region, n (%) | ||||
| Central/Eastern Europe | 73 (47.7) | 21 (50.0) | 7 (87.5) | 101 (49.8) |
| Western Europe | 35 (22.9) | 12 (28.6) | 0 | 47 (23.2) |
| North America | 37 (24.2) | 5 (11.9) | 1 (12.5) | 43 (21.2) |
| Latin America | 5 (3.3) | 2 (4.8) | 0 | 7 (3.4) |
| Asia | 2 (1.3) | 1 (2.4) | 0 | 3 (1.5) |
| Israel | 1 (0.7) | 1 (2.4) | 0 | 2 (1.0) |
| Race,† n (%) | ||||
| White | 141 (92.2) | 37 (88.1) | 7 (87.5) | 185 (91.1) |
| Asian | 6 (3.9) | 1 (2.4) | 0 | 7 (3.4) |
| Black or African American | 4 (2.6) | 3 (7.1) | 0 | 7 (3.4) |
| Multiple | 1 (0.7) | 1 (2.4) | 1 (12.5) | 3 (1.5) |
| Body mass index, mean (SD), kg/m2 | 24.7 (5.4) | 17.8 (2.4) | 16.1 (1.2) | 22.9 (5.7) |
| eGFR,‡ mean (SD), mL/min/1.73 m2 | 101.3 (26.6) | 128.5 (25.2) | 125.6 (15.7) | 107.8 (28.3) |
| Source of patients | ||||
| Chronically anticoagulated children newly exposed to dabigatran | 82 (53.6) | 26 (61.9) | 7 (87.5) | 115 (56.7) |
| Rollover patients treated with dabigatran in the DIVERSITY trial | 46 (30.1) | 12 (28.6) | 1 (12.5) | 59 (29.1) |
| Rollover patients treated with SOC in the DIVERSITY trial | 25 (16.3) | 4 (9.5) | 0 | 29 (14.3) |
| Prior anticoagulation treatment,§n (%) | ||||
| LMWH | 116 (75.8) | 29 (69.0) | 7 (87.5) | 152 (74.9) |
| VKA | 61 (39.9) | 11 (26.2) | 1 (12.5) | 73 (36.0) |
| Unfractionated heparin | 46 (30.1) | 10 (23.8) | 2 (25.0) | 58 (28.6) |
| Direct oral anticoagulant|| | 46 (30.1) | 12 (28.6) | 1 (12.5) | 59 (29.1) |
| Fondaparinux | 1 (0.7) | 0 | 0 | 1 (0.5) |
| Other | 14 (9.2) | 1 (2.4) | 0 | 15 (7.4) |
| Most recent venous thromboembolic event,¶n (%) | ||||
| DVT (other than central-line and cerebral venous thrombosis) | 126 (82.4) | 24 (57.1) | 5 (62.5) | 155 (76.4) |
| Pulmonary embolism | 19 (12.4) | 1 (2.4) | 0 | 20 (9.9) |
| Central-line thrombosis | 2 (1.3) | 3 (7.1) | 2 (25.0) | 7 (3.4) |
| Cerebral venous thrombosis and/or sinus thrombosis | 8 (5.2) | 14 (33.3) | 1 (12.5) | 23 (11.3) |
| DVT (other than central-line and cerebral venous thrombosis) or central-line thrombosis | 128 (83.7) | 27 (64.3) | 7 (87.5) | 162 (79.8) |
| Risk factors for VTE, n (%) | ||||
| 1 prespecified# risk factor | 93 (60.8) | 28 (66.7) | 5 (62.5) | 126 (62.1) |
| ≥2 prespecified# risk factor | 50 (32.7) | 9 (21.4) | 2 (25.0) | 61 (30.0) |
| Other risk factors requiring further anticoagulation** | 10 (6.5) | 5 (11.9) | 1 (12.5) | 16 (7.9) |
| Dabigatran | Total, N = 203 | |||
|---|---|---|---|---|
| 12 to <18 y, n = 153 | 2 to <12 y, n = 42 | 0 to <2 y, n = 8 | ||
| Dabigatran formulation,*n (%) | ||||
| Capsules | 153 (100.0) | 18 (42.9) | 0 | 171 (84.2) |
| Pellets | 0 | 24 (57.1) | 8 (100.0) | 32 (15.8) |
| Age, mean (SD), y | 15.1 (1.6) | 7.0 (3.0) | 0.6 (0.5) | 12.8 (4.5) |
| Male, n (%) | 87 (56.9) | 21 (50.0) | 5 (62.5) | 113 (55.7) |
| Region, n (%) | ||||
| Central/Eastern Europe | 73 (47.7) | 21 (50.0) | 7 (87.5) | 101 (49.8) |
| Western Europe | 35 (22.9) | 12 (28.6) | 0 | 47 (23.2) |
| North America | 37 (24.2) | 5 (11.9) | 1 (12.5) | 43 (21.2) |
| Latin America | 5 (3.3) | 2 (4.8) | 0 | 7 (3.4) |
| Asia | 2 (1.3) | 1 (2.4) | 0 | 3 (1.5) |
| Israel | 1 (0.7) | 1 (2.4) | 0 | 2 (1.0) |
| Race,† n (%) | ||||
| White | 141 (92.2) | 37 (88.1) | 7 (87.5) | 185 (91.1) |
| Asian | 6 (3.9) | 1 (2.4) | 0 | 7 (3.4) |
| Black or African American | 4 (2.6) | 3 (7.1) | 0 | 7 (3.4) |
| Multiple | 1 (0.7) | 1 (2.4) | 1 (12.5) | 3 (1.5) |
| Body mass index, mean (SD), kg/m2 | 24.7 (5.4) | 17.8 (2.4) | 16.1 (1.2) | 22.9 (5.7) |
| eGFR,‡ mean (SD), mL/min/1.73 m2 | 101.3 (26.6) | 128.5 (25.2) | 125.6 (15.7) | 107.8 (28.3) |
| Source of patients | ||||
| Chronically anticoagulated children newly exposed to dabigatran | 82 (53.6) | 26 (61.9) | 7 (87.5) | 115 (56.7) |
| Rollover patients treated with dabigatran in the DIVERSITY trial | 46 (30.1) | 12 (28.6) | 1 (12.5) | 59 (29.1) |
| Rollover patients treated with SOC in the DIVERSITY trial | 25 (16.3) | 4 (9.5) | 0 | 29 (14.3) |
| Prior anticoagulation treatment,§n (%) | ||||
| LMWH | 116 (75.8) | 29 (69.0) | 7 (87.5) | 152 (74.9) |
| VKA | 61 (39.9) | 11 (26.2) | 1 (12.5) | 73 (36.0) |
| Unfractionated heparin | 46 (30.1) | 10 (23.8) | 2 (25.0) | 58 (28.6) |
| Direct oral anticoagulant|| | 46 (30.1) | 12 (28.6) | 1 (12.5) | 59 (29.1) |
| Fondaparinux | 1 (0.7) | 0 | 0 | 1 (0.5) |
| Other | 14 (9.2) | 1 (2.4) | 0 | 15 (7.4) |
| Most recent venous thromboembolic event,¶n (%) | ||||
| DVT (other than central-line and cerebral venous thrombosis) | 126 (82.4) | 24 (57.1) | 5 (62.5) | 155 (76.4) |
| Pulmonary embolism | 19 (12.4) | 1 (2.4) | 0 | 20 (9.9) |
| Central-line thrombosis | 2 (1.3) | 3 (7.1) | 2 (25.0) | 7 (3.4) |
| Cerebral venous thrombosis and/or sinus thrombosis | 8 (5.2) | 14 (33.3) | 1 (12.5) | 23 (11.3) |
| DVT (other than central-line and cerebral venous thrombosis) or central-line thrombosis | 128 (83.7) | 27 (64.3) | 7 (87.5) | 162 (79.8) |
| Risk factors for VTE, n (%) | ||||
| 1 prespecified# risk factor | 93 (60.8) | 28 (66.7) | 5 (62.5) | 126 (62.1) |
| ≥2 prespecified# risk factor | 50 (32.7) | 9 (21.4) | 2 (25.0) | 61 (30.0) |
| Other risk factors requiring further anticoagulation** | 10 (6.5) | 5 (11.9) | 1 (12.5) | 16 (7.9) |
DVT, deep vein thrombosis; eGFR, estimated glomerular filtration rate; SD, standard deviation.
One adolescent was not treated.
Missing data for 1 adolescent.
Calculated using the Schwartz formula.
Nonanticoagulation therapy was used to treat the most recent VTE in 5 adolescents, 2 children aged 2 to <12 years, and 1 child aged from >3 months to <2 years.
Rollover patients treated with dabigatran in the DIVERSITY trial.
Patients could be assessed with >1 type of most recent VTE.
Defined as inherited thrombophilia, short bowel syndrome, congenital nephrotic syndrome, inflammatory bowel disease, recent immobilization, presence of central venous/arterial line or catheter, total parental nutrition, systemic lupus erythematosus, systemic sclerosis or inflammatory vasculopathies, recurrent idiopathic (unprovoked) VTE, structural venous abnormality, antiphospholipid and/or lupus antibodies.
As assessed by investigators, not within prespecified categories of risk factors.
Information on previous VTE and baseline medical conditions with increased risk of thrombosis was available for 199 of 203 patients. Prior confirmed VTE (before the index [most recent] VTE event in this trial) was reported by 36 of 199 children (18.1%) (range of 2-6 confirmed VTE events including the most recent event) (Table 2). Previous VTE was reported as being unprovoked in 25 of 36 (69.4%) and provoked in 13 of 36 children (36.1%). There were 35 of 199 children (17.6%) who had PTS at baseline. The most common medical conditions with increased risk of thrombosis were congenital heart disease (12 of 199 [6.0%]), hematologic cancer (11 of 199 [5.5%]), and the presence of a central venous line or catheter (11 of 199 [5.5%]). Inherited thrombophilia was reported in 91 of 203 children (44.8%); of these, the following disorders were reported: factor V Leiden mutation in 34 (16.7%), prothrombin mutation in 17 (8.4%), antithrombin deficiency in 20 (9.9%), protein C/S deficiency in 23 (11.3%), and other coagulation disorders in 23 (11.3%). Of other conditions requiring secondary VTE prophylaxis, recurrent unprovoked VTE was the most common, reported for 29 of 203 children (14.3%).
Baseline risk factors for VTE by age strata
| Dabigatran | Total | |||
|---|---|---|---|---|
| 12 to <18 y | 2 to <12 y | 0 to <2 y | ||
| Medical history of previous thromboembolic events | n = 150 | n = 41 | n = 8 | N = 199* |
| History of prior VTE event (before the index VTE event in this trial), n (%) | ||||
| Yes | 28 (18.7) | 6 (14.6) | 2 (25.0) | 36 (18.1) |
| No. of prior confirmed VTE events† | ||||
| 2 | 9 (6.0) | 1 (2.4) | 0 | 10 (5.0) |
| 3 | 13 (8.7) | 4 (9.8) | 1 (12.5) | 18 (9.0) |
| 4 | 4 (2.7) | 0 | 0 | 4 (2.0) |
| 5 | 1 (0.7) | 0 | 0 | 1 (0.5) |
| 6 | 1 (0.7) | 1 (2.4) | 1 (12.5) | 3 (1.5) |
| No | 122 (81.3) | 35 (85.4) | 6 (75.0) | 163 (81.9) |
| Previous VTE,‡ n (%) | n = 28 | n = 6 | n = 2 | n = 36 |
| Unprovoked | 21 (75.0) | 3 (50.0) | 1 (50.0) | 25 (69.4) |
| Provoked | 7 (25.0) | 4 (66.7) | 2 (100.0) | 13 (36.1) |
| Postthrombotic syndrome,§ n (%) | 29 (19.3) | 5 (12.2) | 1 (12.5) | 35 (17.6) |
| Medical conditions/circumstances with increased risk of thrombosis, n (%) | n = 150 | n = 41 | n = 8 | N = 199* |
| Congenital heart disease | 6 (4.0) | 3 (7.3) | 3 (37.5) | 12 (6.0) |
| Hematologic cancer | 4 (2.7) | 7 (17.1) | 0 | 11 (5.5) |
| Presence of central venous line | 3 (2.0) | 4 (9.8) | 4 (50.0) | 11 (5.5) |
| Recent immobilization|| | 7 (4.7) | 0 | 0 | 7 (3.5) |
| Any history of solid cancer | 1 (0.7) | 2 (4.9) | 1 (12.5) | 4 (2.0) |
| Presence of other venous or arterial catheter | 2 (1.3) | 1 (2.4) | 0 | 3 (1.5) |
| Hypertension | 3 (2.0) | 0 | 0 | 3 (1.5) |
| Heart failure | 0 | 1 (2.4) | 1 (12.5) | 2 (1.0) |
| History of stroke or transient ischemic attack | 0 | 1 (2.4) | 0 | 1 (0.5) |
| Liver disease (currently not active) | 0 | 0 | 1 (12.5) | 1 (0.5) |
| History of major or clinically relevant bleeding event | 0 | 1 (2.4) | 0 | 1 (0.5) |
| Total parenteral nutrition dependency | 1 (0.7) | 0 | 0 | 1 (0.5) |
| Clinical risk factors requiring secondary VTE prevention, n (%) | n = 153 | n = 42 | n = 8 | N = 203 |
| Inherited thrombophilia¶,# | 79 (51.6) | 11 (26.2) | 1 (12.5) | 91 (44.8) |
| Factor V Leiden mutation** | 32 (20.9) | 2 (4.8) | 0 | 34 (16.7) |
| Prothrombin mutation†† | 15 (9.8) | 2 (4.8) | 0 | 17 (8.4) |
| Antithrombin deficiency | 16 (10.5) | 4 (9.5) | 0 | 20 (9.9) |
| Protein S/C deficiency | 19 (12.4) | 4 (9.5) | 0 | 23 (11.3) |
| Other‡‡ | 20 (13.1) | 2 (4.8) | 1 (12.5) | 23 (11.3) |
| 2 or more thrombophilia conditions | 26 (17.0) | 4 (9.5) | 0 | 30 (14.8) |
| Congenital nephrotic syndrome | 0 | 1 (2.4) | 0 | 1 (0.5) |
| Other conditions requiring secondary VTE prophylaxis, n (%) | n = 153 | n = 42 | n = 8 | N = 203 |
| Antiphospholipid antibodies and/or lupus antibodies | 18 (11.8) | 1 (2.4) | 1 (12.5) | 20 (9.9) |
| Recurrent unprovoked VTE | 21 (13.7) | 7 (16.7) | 1 (12.5) | 29 (14.3) |
| Structural venous abnormality§§ | 20 (13.1) | 6 (14.3) | 0 | 26 (12.8) |
| Any other risk factor requiring secondary prophylaxis | 47 (30.7) | 18 (42.9) | 6 (75.0) | 71 (35.0) |
| Dabigatran | Total | |||
|---|---|---|---|---|
| 12 to <18 y | 2 to <12 y | 0 to <2 y | ||
| Medical history of previous thromboembolic events | n = 150 | n = 41 | n = 8 | N = 199* |
| History of prior VTE event (before the index VTE event in this trial), n (%) | ||||
| Yes | 28 (18.7) | 6 (14.6) | 2 (25.0) | 36 (18.1) |
| No. of prior confirmed VTE events† | ||||
| 2 | 9 (6.0) | 1 (2.4) | 0 | 10 (5.0) |
| 3 | 13 (8.7) | 4 (9.8) | 1 (12.5) | 18 (9.0) |
| 4 | 4 (2.7) | 0 | 0 | 4 (2.0) |
| 5 | 1 (0.7) | 0 | 0 | 1 (0.5) |
| 6 | 1 (0.7) | 1 (2.4) | 1 (12.5) | 3 (1.5) |
| No | 122 (81.3) | 35 (85.4) | 6 (75.0) | 163 (81.9) |
| Previous VTE,‡ n (%) | n = 28 | n = 6 | n = 2 | n = 36 |
| Unprovoked | 21 (75.0) | 3 (50.0) | 1 (50.0) | 25 (69.4) |
| Provoked | 7 (25.0) | 4 (66.7) | 2 (100.0) | 13 (36.1) |
| Postthrombotic syndrome,§ n (%) | 29 (19.3) | 5 (12.2) | 1 (12.5) | 35 (17.6) |
| Medical conditions/circumstances with increased risk of thrombosis, n (%) | n = 150 | n = 41 | n = 8 | N = 199* |
| Congenital heart disease | 6 (4.0) | 3 (7.3) | 3 (37.5) | 12 (6.0) |
| Hematologic cancer | 4 (2.7) | 7 (17.1) | 0 | 11 (5.5) |
| Presence of central venous line | 3 (2.0) | 4 (9.8) | 4 (50.0) | 11 (5.5) |
| Recent immobilization|| | 7 (4.7) | 0 | 0 | 7 (3.5) |
| Any history of solid cancer | 1 (0.7) | 2 (4.9) | 1 (12.5) | 4 (2.0) |
| Presence of other venous or arterial catheter | 2 (1.3) | 1 (2.4) | 0 | 3 (1.5) |
| Hypertension | 3 (2.0) | 0 | 0 | 3 (1.5) |
| Heart failure | 0 | 1 (2.4) | 1 (12.5) | 2 (1.0) |
| History of stroke or transient ischemic attack | 0 | 1 (2.4) | 0 | 1 (0.5) |
| Liver disease (currently not active) | 0 | 0 | 1 (12.5) | 1 (0.5) |
| History of major or clinically relevant bleeding event | 0 | 1 (2.4) | 0 | 1 (0.5) |
| Total parenteral nutrition dependency | 1 (0.7) | 0 | 0 | 1 (0.5) |
| Clinical risk factors requiring secondary VTE prevention, n (%) | n = 153 | n = 42 | n = 8 | N = 203 |
| Inherited thrombophilia¶,# | 79 (51.6) | 11 (26.2) | 1 (12.5) | 91 (44.8) |
| Factor V Leiden mutation** | 32 (20.9) | 2 (4.8) | 0 | 34 (16.7) |
| Prothrombin mutation†† | 15 (9.8) | 2 (4.8) | 0 | 17 (8.4) |
| Antithrombin deficiency | 16 (10.5) | 4 (9.5) | 0 | 20 (9.9) |
| Protein S/C deficiency | 19 (12.4) | 4 (9.5) | 0 | 23 (11.3) |
| Other‡‡ | 20 (13.1) | 2 (4.8) | 1 (12.5) | 23 (11.3) |
| 2 or more thrombophilia conditions | 26 (17.0) | 4 (9.5) | 0 | 30 (14.8) |
| Congenital nephrotic syndrome | 0 | 1 (2.4) | 0 | 1 (0.5) |
| Other conditions requiring secondary VTE prophylaxis, n (%) | n = 153 | n = 42 | n = 8 | N = 203 |
| Antiphospholipid antibodies and/or lupus antibodies | 18 (11.8) | 1 (2.4) | 1 (12.5) | 20 (9.9) |
| Recurrent unprovoked VTE | 21 (13.7) | 7 (16.7) | 1 (12.5) | 29 (14.3) |
| Structural venous abnormality§§ | 20 (13.1) | 6 (14.3) | 0 | 26 (12.8) |
| Any other risk factor requiring secondary prophylaxis | 47 (30.7) | 18 (42.9) | 6 (75.0) | 71 (35.0) |
Medical history of previous VTE was collected starting clinical report form version 2; missing data for 4 children who were assessed with clinical report form version 1.
Includes the most recent VTE event.
Children may be counted in >1 category. Percentages based on the number of patients with a history of prior VTE events. Missing data for 163 children.
Missing data for 1 adolescent.
Illness requiring bed rest or involving paralysis.
The number of children with ≥1 of the conditions listed.
Missing data for 2 adolescents and 2 children ages 2 to <12 years.
Gln506 (rs6025), hetero- or homozygous.
G20210A mutation.
Other coagulation disorders/thrombophilias including: MTHFR mutation, methionine synthase reductase mutation, plasminogen activator inhibitor 4G/5G polymorphism, factor XII deficiency, integrin A2 mutation, hyperhomocysteinemia, fibrinogen mutation, glycoprotein IA mutation, glycoprotein IIIA mutation, factor VIII elevation.
Structurally abnormal venous system, for example, inferior vena cava malformation, Paget-Schroetter disease (thoracic outlet syndrome), May-Thurner syndrome (iliac vein compression syndrome).
Primary end points
Overall, 2 of 203 children (1.0%) experienced recurrent VTE, 1 within 3 months of treatment and the other within 6 months of treatment (Table 3; Figure 2A). Both were female adolescents who had had deep vein thrombosis (n = 1) and pulmonary embolism (n = 1) as their most recent VTE event. Clinical risk factors that required secondary VTE protection were methylenetetrahydrofolate reductase (MTHFR) A1298C homozygous mutation in 1 female patient whose most recent VTE event was 3.5 months prior. In the other female patient, whose most recent VTE event was 26 months prior, they were MTHFR A1298C, plasminogen activator inhibitor-1 46156 heterozygote mutation, and protein S deficiency. The probability of freedom from recurrent VTE at 12 months across all age strata during the on-treatment period was 0.988 (95% confidence interval [CI], 0.951-0.997). Consistent results were observed for the full study period: on-treatment plus follow-up (probability of freedom from recurrent VTE at 12 months, 0.952; CI, 0.900-0.977).
On-treatment VTEs, PTS, bleeding events, and fatal events at 12 months by age strata (treated set)
| Dabigatran | Total, N = 203 | |||
|---|---|---|---|---|
| 12 to <18 y, n = 153 | 2 to <12 y, n = 42 | 0 to <2 y, n = 8 | ||
| Recurrent VTE event, n (%) | 2 (1.3) | 0 | 0 | 2 (1.0) |
| Subgroup analysis,* n/N (%) | ||||
| Sex | ||||
| Male | 0/87 (0.0) | 0/21 (0.0) | 0/5 (0.0) | 0/113 (0.0) |
| Female | 2/66 (3.0) | 0/21 (0.0) | 0/3 (0.0) | 2/90 (2.2) |
| PTS,†n (%) | 2/128 (1.6) | 0/27 (0.0) | 0/7 (0.0) | 2/162 (1.2) |
| Subgroup analysis,* n/N (%) | ||||
| Sex | ||||
| Male | 2/80 (2.5) | 0/12 (0.0) | 0/5 (0.0) | 2/97 (2.1) |
| Female | 0/48 (0.0) | 0/15 (0.0) | 0/2 (0.0) | 0/65 (0.0) |
| Bleeding events, n (%) | 37 (24.2) | 2 (4.8) | 1 (12.5) | 40 (19.7) |
| Major | 3 (2.0) | 0 | 0 | 3 (1.5) |
| Clinically relevant non-major | 1 (0.7) | 1 (2.4) | 0 | 2 (1.0) |
| Minor | 34 (22.2) | 2 (4.8) | 1 (12.5) | 37 (18.2) |
| All-cause death, n (%) | 0 | 0 | 0 | 0 |
| Dabigatran | Total, N = 203 | |||
|---|---|---|---|---|
| 12 to <18 y, n = 153 | 2 to <12 y, n = 42 | 0 to <2 y, n = 8 | ||
| Recurrent VTE event, n (%) | 2 (1.3) | 0 | 0 | 2 (1.0) |
| Subgroup analysis,* n/N (%) | ||||
| Sex | ||||
| Male | 0/87 (0.0) | 0/21 (0.0) | 0/5 (0.0) | 0/113 (0.0) |
| Female | 2/66 (3.0) | 0/21 (0.0) | 0/3 (0.0) | 2/90 (2.2) |
| PTS,†n (%) | 2/128 (1.6) | 0/27 (0.0) | 0/7 (0.0) | 2/162 (1.2) |
| Subgroup analysis,* n/N (%) | ||||
| Sex | ||||
| Male | 2/80 (2.5) | 0/12 (0.0) | 0/5 (0.0) | 2/97 (2.1) |
| Female | 0/48 (0.0) | 0/15 (0.0) | 0/2 (0.0) | 0/65 (0.0) |
| Bleeding events, n (%) | 37 (24.2) | 2 (4.8) | 1 (12.5) | 40 (19.7) |
| Major | 3 (2.0) | 0 | 0 | 3 (1.5) |
| Clinically relevant non-major | 1 (0.7) | 1 (2.4) | 0 | 2 (1.0) |
| Minor | 34 (22.2) | 2 (4.8) | 1 (12.5) | 37 (18.2) |
| All-cause death, n (%) | 0 | 0 | 0 | 0 |
Missing data for 1 adolescent.
Calculated over the number of patients with deep vein thrombosis (other than central-line and cerebral venous thrombosis) or central-line thrombosis.
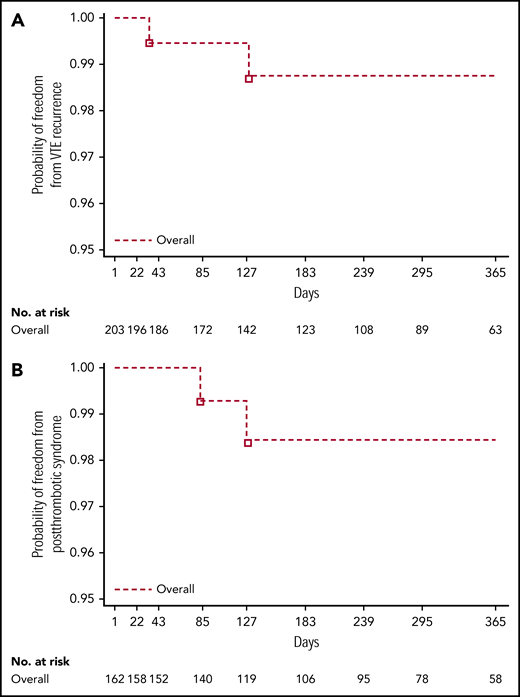
Kaplan-Meier curves for all age strata. Combined (on-treatment) for time to recurrent VTE (A) and postthrombotic syndrome (adjudicated data from the treated set) (B).
Kaplan-Meier curves for all age strata. Combined (on-treatment) for time to recurrent VTE (A) and postthrombotic syndrome (adjudicated data from the treated set) (B).
The probability of freedom from bleeding at 12 months during the on-treatment period was 0.752 (95% CI, 0.672-0.815). Consistent results were observed for the full study period: on-treatment plus follow-up (probability of freedom from bleeding at 12 months, 0.771; CI, 0.699-0.827). Of the 40 of 203 children (19.7%) reporting bleeding events, 1.5% (3 of 203) were MBEs, 1.0% (2 of 203) were CRNMBEs, and 18.2% (37 of 203) were minor bleeding events (Table 3). The location of investigator-reported on-treatment bleeding events with dabigatran is shown in supplemental Table 2. Of the 3 children reporting MBEs, 1 was a 17-year-old female patient with an MBE originating from a venous varix in the right leg (treated with idarucizumab [Praxbind] to reverse dabigatran in the context of a clinical trial [NCT02815670]); 1 was a 16-year-old male patient who experienced an extrapleural hematoma following surgery, 3 days after a temporary interruption of dabigatran for planned surgery; and 1 was a 17-year-old male patient with thrombophilia and positive tests for homocysteinemia, high levels of lupus anticoagulant, and anticardiolipin antibodies who presented with an episode of hemoptysis (treatment with dabigatran was switched to another anticoagulant). This patient experienced a pulmonary embolism with a fatal outcome 6 days after stopping dabigatran treatment. For the 2 children reporting CRNMBEs, 1 was a 17-year-old male patient who experienced a cut in a finger that required medical/surgical intervention (stitches) to achieve hemostasis, and 1 was a 10-year-old female patient who presented with heavy menses after starting her menstrual cycle. There were no deaths while patients were on treatment.
Secondary end points
In total, PTS was newly reported in 2 male adolescents of 162 patients (1.2%) who had DVT or central-line thrombosis as their most recent VTE (Table 3; Figure 2B), 1 within 3 months and the other within 3 to 6 months of treatment. The probability of freedom from newly diagnosed PTS at 12 months across all age strata during the on-treatment period was 0.985 (95% CI, 0.939-0.996). Consistent results were observed for the full study period: on-treatment plus follow-up (probability of freedom from newly diagnosed PTS at 12 months, 0.980; 95% CI, 0.938-0.993). Clinical risk factors that required secondary VTE protection in these patients were (i) factor V Leiden (heterozygous mutation) and prothrombin mutation (homozygous mutation) in 1 male patient whose most recent VTE event was 3 months prior (modified Villalta score 2) and (ii) factor V Leiden (heterozygous mutation), prothrombin (heterozygous mutation), and complete occlusion of the vena cava in 1 male patient whose most recent VTE event was 11.5 months prior (modified Villalta score 3).
Adverse events were reported by 152 of 203 children (74.9%), with the most common being nasopharyngitis (34 of 203 [16.7%]), headache (33 of 203 [16.3%]), and abdominal pain (21 of 203 [10.3%]) (Table 4). Serious adverse events were experienced by 25 of 203 children (12.3%), and adverse events leading to treatment discontinuation were reported by 12 of 203 children (5.9%).
Summary of adverse events, and adverse events occurring in ≥5% of children in either treatment group (treated set)
| Dabigatran | Total, N = 203 | |||
|---|---|---|---|---|
| 12 to <18 y, n = 153 | 2 to <12 y, n = 42 | 0 to <2 y, n = 8 | ||
| Children with any adverse event, n (%) | 120 (78.4) | 25 (59.5) | 7 (87.5) | 152 (74.9) |
| Drug-related adverse event* | 37 (24.2) | 5 (11.9) | 1 (12.5) | 43 (21.2) |
| Children with serious adverse events,† n (%) | 19 (12.4) | 6 (14.3) | 0 | 25 (12.3) |
| Leading to death | 0 | 0 | 0 | 0 |
| Life-threatening | 2 (1.3) | 0 | 0 | 2 (1.0) |
| Requiring hospitalization | 17 (11.1) | 6 (14.3) | 0 | 23 (11.3) |
| Prolonging hospitalization | 1 (0.7) | 1 (2.4) | 0 | 2 (1.0) |
| Other | 2 (1.3) | 2 (4.8) | 0 | 4 (2.0) |
| Children with adverse events of special interest‡ | 0 | 0 | 0 | 0 |
| Children with adverse events leading to treatment discontinuation | 9 (5.9) | 2 (4.8) | 1 (12.5) | 12 (5.9) |
| Adverse events in ≥5% of children overall, n (%) | ||||
| Nasopharyngitis | 26 (17.0) | 7 (16.7) | 1 (12.5) | 34 (16.7) |
| Drug-related | 0 | 0 | 0 | 0 |
| Serious adverse event | 0 | 0 | 0 | 0 |
| Headache | 27 (17.6) | 6 (14.3) | 0 | 33 (16.3) |
| Drug-related | 2 (1.3) | 1 (2.4) | 0 | 3 (1.5) |
| Serious | 0 | 1 (2.4) | 0 | 1 (0.5) |
| Abdominal pain§ | 19 (12.4) | 2 (4.8) | 0 | 21 (10.3) |
| Drug-related | 5 (3.3) | 2 (4.8) | 0 | 7 (3.4) |
| Serious | 1 (0.7) | 0 | 0 | 1 (0.5) |
| Respiratory tract infection|| | 15 (9.8) | 3 (7.1) | 0 | 18 (8.9) |
| Drug-related | 0 | 0 | 0 | 0 |
| Serious | 1 (0.7) | 0 | 0 | 1 (0.5) |
| Nausea | 13 (8.5) | 3 (7.1) | 0 | 16 (7.9) |
| Drug-related | 5 (3.3) | 2 (4.8) | 0 | 7 (3.4) |
| Serious | 0 | 0 | 0 | 0 |
| Vomiting | 10 (6.5) | 4 (9.5) | 0 | 14 (6.9) |
| Drug-related | 1 (0.7) | 1 (2.4) | 0 | 2 (1.0) |
| Serious | 0 | 0 | 0 | 0 |
| Cough | 9 (5.9) | 4 (9.5) | 1 (12.5) | 14 (6.9) |
| Drug-related | 0 | 0 | 0 | 0 |
| Serious | 0 | 0 | 0 | 0 |
| Dyspepsia | 13 (8.5) | 0 | 0 | 13 (6.4) |
| Drug-related | 9 (5.9) | 0 | 0 | 9 (4.4) |
| Serious | 0 | 0 | 0 | 0 |
| Pyrexia | 11 (7.2) | 2 (4.8) | 0 | 13 (6.4) |
| Drug-related | 0 | 0 | 0 | 0 |
| Serious | 0 | 0 | 0 | 0 |
| Diarrhea | 12 (7.8) | 1 (2.4) | 0 | 13 (6.4) |
| Drug-related | 2 (1.3) | 0 | 0 | 2 (1.0) |
| Serious | 0 | 0 | 0 | 0 |
| Pain in extremity | 11 (7.2) | 2 (4.8) | 0 | 13 (6.4) |
| Drug-related | 0 | 0 | 0 | 0 |
| Serious | 1 (0.7) | 0 | 0 | 1 (0.5) |
| Epistaxis | 10 (6.5) | 1 (2.4) | 1 (12.5) | 12 (5.9) |
| Drug-related | 5 (3.3) | 1 (2.4) | 1 (12.5) | 7 (3.4) |
| Serious | 0 | 0 | 0 | 0 |
| Dabigatran | Total, N = 203 | |||
|---|---|---|---|---|
| 12 to <18 y, n = 153 | 2 to <12 y, n = 42 | 0 to <2 y, n = 8 | ||
| Children with any adverse event, n (%) | 120 (78.4) | 25 (59.5) | 7 (87.5) | 152 (74.9) |
| Drug-related adverse event* | 37 (24.2) | 5 (11.9) | 1 (12.5) | 43 (21.2) |
| Children with serious adverse events,† n (%) | 19 (12.4) | 6 (14.3) | 0 | 25 (12.3) |
| Leading to death | 0 | 0 | 0 | 0 |
| Life-threatening | 2 (1.3) | 0 | 0 | 2 (1.0) |
| Requiring hospitalization | 17 (11.1) | 6 (14.3) | 0 | 23 (11.3) |
| Prolonging hospitalization | 1 (0.7) | 1 (2.4) | 0 | 2 (1.0) |
| Other | 2 (1.3) | 2 (4.8) | 0 | 4 (2.0) |
| Children with adverse events of special interest‡ | 0 | 0 | 0 | 0 |
| Children with adverse events leading to treatment discontinuation | 9 (5.9) | 2 (4.8) | 1 (12.5) | 12 (5.9) |
| Adverse events in ≥5% of children overall, n (%) | ||||
| Nasopharyngitis | 26 (17.0) | 7 (16.7) | 1 (12.5) | 34 (16.7) |
| Drug-related | 0 | 0 | 0 | 0 |
| Serious adverse event | 0 | 0 | 0 | 0 |
| Headache | 27 (17.6) | 6 (14.3) | 0 | 33 (16.3) |
| Drug-related | 2 (1.3) | 1 (2.4) | 0 | 3 (1.5) |
| Serious | 0 | 1 (2.4) | 0 | 1 (0.5) |
| Abdominal pain§ | 19 (12.4) | 2 (4.8) | 0 | 21 (10.3) |
| Drug-related | 5 (3.3) | 2 (4.8) | 0 | 7 (3.4) |
| Serious | 1 (0.7) | 0 | 0 | 1 (0.5) |
| Respiratory tract infection|| | 15 (9.8) | 3 (7.1) | 0 | 18 (8.9) |
| Drug-related | 0 | 0 | 0 | 0 |
| Serious | 1 (0.7) | 0 | 0 | 1 (0.5) |
| Nausea | 13 (8.5) | 3 (7.1) | 0 | 16 (7.9) |
| Drug-related | 5 (3.3) | 2 (4.8) | 0 | 7 (3.4) |
| Serious | 0 | 0 | 0 | 0 |
| Vomiting | 10 (6.5) | 4 (9.5) | 0 | 14 (6.9) |
| Drug-related | 1 (0.7) | 1 (2.4) | 0 | 2 (1.0) |
| Serious | 0 | 0 | 0 | 0 |
| Cough | 9 (5.9) | 4 (9.5) | 1 (12.5) | 14 (6.9) |
| Drug-related | 0 | 0 | 0 | 0 |
| Serious | 0 | 0 | 0 | 0 |
| Dyspepsia | 13 (8.5) | 0 | 0 | 13 (6.4) |
| Drug-related | 9 (5.9) | 0 | 0 | 9 (4.4) |
| Serious | 0 | 0 | 0 | 0 |
| Pyrexia | 11 (7.2) | 2 (4.8) | 0 | 13 (6.4) |
| Drug-related | 0 | 0 | 0 | 0 |
| Serious | 0 | 0 | 0 | 0 |
| Diarrhea | 12 (7.8) | 1 (2.4) | 0 | 13 (6.4) |
| Drug-related | 2 (1.3) | 0 | 0 | 2 (1.0) |
| Serious | 0 | 0 | 0 | 0 |
| Pain in extremity | 11 (7.2) | 2 (4.8) | 0 | 13 (6.4) |
| Drug-related | 0 | 0 | 0 | 0 |
| Serious | 1 (0.7) | 0 | 0 | 1 (0.5) |
| Epistaxis | 10 (6.5) | 1 (2.4) | 1 (12.5) | 12 (5.9) |
| Drug-related | 5 (3.3) | 1 (2.4) | 1 (12.5) | 7 (3.4) |
| Serious | 0 | 0 | 0 | 0 |
Investigator defined.
A child could be counted in >1 category.
Protocol-defined adverse events of special interest were elevated aspartate aminotransferase and/or alanine aminotransferase more than threefold upper limit of normal combined with an elevation of total bilirubin more than twofold upper limit of normal measured in the same blood draw sample, and an at least twofold increase in creatinine from baseline levels that is above the upper limit of normal.
Includes children with upper abdominal pain.
Includes children with upper respiratory tract infection and viral upper respiratory tract infection.
At end of treatment, according to investigators, 124 of 126 children (98.4%) and 16 of 17 children (94.1%) had good or satisfactory acceptance of capsules and pellets, respectively, and were able to take them very often or all of the time (124 of 126 [98.4%] and 16 of 17 [94.1%] for capsules and pellets, respectively) (supplemental Table 3). Overall, dabigatran geometric mean trough exposure over all visits was 88.8 ng/mL (42.6); across age strata it was 96.5 ng/mL (34.8) for children aged 12 to <18 years (n = 153), 74.7 ng/mL (37.1) for those aged 2 to <12 years (n = 38), and 40.9 ng/mL (97.1) for those aged from >3 months to <2 years (n = 8) (supplemental Table 4). Pharmacokinetic/pharmacodynamic curves showed a linear relationship between total dabigatran plasma concentration and dTT and ECT, and a nonlinear relationship between total dabigatran plasma concentrations and aPTT (Figure 3). During the on-treatment period, 26.1% of patients had a dose adjustment (increase or decrease of dose).
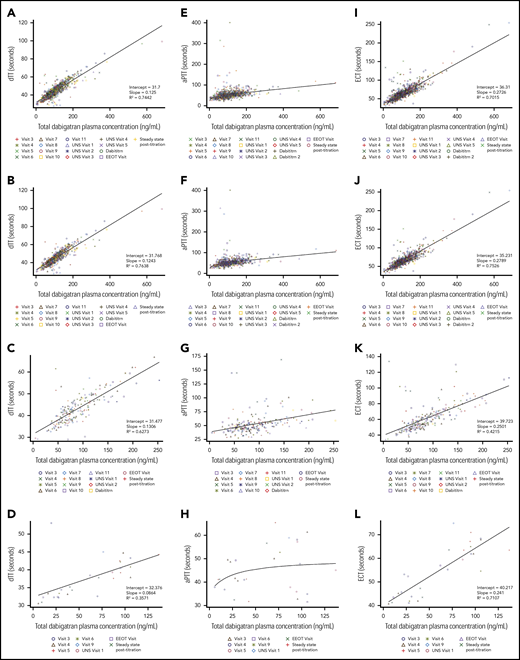
Pharmacokinetic-pharmacodynamic relationship curves at trough sampling times. For dabigatran PK and dTT (A, overall; B, aged 12 to <18 years; C, aged 2 to <12 years; D, aged from >3 months to <2 years), aPTT (E, overall; F, aged 12 to <18 years; G, aged 2 to <12 years; H, aged from >3 months to <2 years), and ECT (I, overall; J, aged 12 to <18 years; K, aged 2 to <12 years; L, aged from >3 months to <2 years) by age group. aPTT, activated partial thrombin time; Dabititrn, dabigatran titration; EEOT, early end of treatment; UNS, unscheduled.
Pharmacokinetic-pharmacodynamic relationship curves at trough sampling times. For dabigatran PK and dTT (A, overall; B, aged 12 to <18 years; C, aged 2 to <12 years; D, aged from >3 months to <2 years), aPTT (E, overall; F, aged 12 to <18 years; G, aged 2 to <12 years; H, aged from >3 months to <2 years), and ECT (I, overall; J, aged 12 to <18 years; K, aged 2 to <12 years; L, aged from >3 months to <2 years) by age group. aPTT, activated partial thrombin time; Dabititrn, dabigatran titration; EEOT, early end of treatment; UNS, unscheduled.
Discussion
This trial is the first to describe clinical outcomes in children aged <18 years being treated with dabigatran etexilate for secondary VTE prevention. We observed a low frequency of recurrent VTEs and few MBEs or CRNMBEs during dabigatran treatment in this setting. The rate of newly diagnosed PTS with dabigatran treatment in the study was 2 of 162 (1.2%). Using an age- and body-weight–adjusted dosing algorithm, dabigatran plasma trough exposure was similar to that observed in adults.16 In addition, the dabigatran pharmacokinetic/pharmacodynamic relationship seen in this pediatric population was similar to that seen in the adult setting.23
In terms of antithrombotic effect, the DIVERSITY trial has shown similar efficacy for dabigatran vs SOC when treating children aged <18 years with acute VTE, as well as the suitability of a pediatric dabigatran-dosing algorithm.24 In that trial, there was a slightly higher frequency of recurrent VTE in children being treated with dabigatran for acute VTE (3.8%) than in this trial (1.0%).24 This difference likely reflects the time since the index VTE event occurred, accompanied by a higher prothrombotic state during their acute VTE setting.
Our study population was composed of ∼20% of patients with the so-called high-risk inherited thrombophilia (protein S/C or antithrombin deficiency) and an additional 10% of patients with antiphospholipid syndrome (APS). In comparison, European investigators analyzed a multicenter German-Israeli database to study the impact of genetically confirmed, high-risk, inherited thrombophilia on recurrent VTE in children and adults after cessation of anticoagulation.25 A cumulative incidence of thrombosis recurrence of ∼4% was observed at 12 months of follow-up among pediatric patients.25 A different analysis of the same database suggests that these patients may have been off anticoagulation between 3 and 9 months after their event,26 which could explain their higher frequency of thrombosis recurrence. Additionally, 2 pediatric studies on APS in patients with arterial and venous thrombosis reported recurrence of thrombosis rates of 19% and 29% with mean follow-up periods of 6.1 and 5.7 years, respectively.27,28 Although the follow-up duration of those studies was longer than in our study, and the participants of those may not match our study population, these comparisons contribute to placing the overall VTE recurrence rate reported herein in the context of the literature. To address the issue of lupus anticoagulants and direct oral anticoagulants (DOACs) in adult patients with high-risk, triple-positive APS, a lack of benefit and increased thrombotic risk has been reported following treatment with the direct factor Xa inhibitor rivaroxaban compared with warfarin,29 raising the question of whether all DOACs should be avoided in this setting. However, as dabigatran has a different mode of action and directly inhibits thrombin, dabigatran might potentially lead to a more beneficial outcome in such patients. Indeed, a post hoc pooled analysis of the RE-COVER/RE-COVER II and RE-MEDY trials found no significant differences in efficacy and safety between dabigatran and warfarin in adult patients with APS.30 Of note, APS-positive patients comprised a small number of subjects enrolled (71 treated with dabigatran vs 80 treated with warfarin), and a distinction between high-risk or regular APS status was not included.30 At present, prospective data regarding the efficacy and safety of dabigatran in the high-risk, triple-positive APS setting are lacking, and clinical trials are required.
Importantly, our trial results compare favorably to prior reports of VTE recurrence in the pediatric population. In the REVIVE trial, an open-label, pediatric, randomized, controlled study in which the LMWH reviparin was compared with unfractionated heparin/VKA to treat VTE, the rate of venous thrombosis recurrence or death of patients treated with LMWH was 5.6%.31 Similarly, a meta-analysis of LMWH pediatric studies reported pooled incidence rates for the development of recurrent VTE on LMWH secondary prophylaxis as being 5.2%,32 whereas other LMWH pediatric dose-finding studies have reported VTE recurrence rates of 3%.33
In both trials, the frequency of any bleeding events on treatment with dabigatran was similar (19.7% in this trial and 19.9% in DIVERSITY), with a low incidence of MBEs in both studies (1.5% in this trial and 2.6% in DIVERSITY). Our bleeding complications align with results in the pediatric literature. The REVIVE trial with the LMWH reviparin reported a major bleeding rate of 5.6%.31 Likewise, a meta-analysis of LMWH pediatric studies reported a pooled incidence rate for major bleeding of 1.8% and 6.5% for once-daily and twice-daily administration, respectively.32
In relation to newly diagnosed PTS, the incidence of 1.2% (all cases being possible mild ones) raises the concern as to whether these data were evaluated prematurely. Of note, the last official recommendation from the International Society on Thrombosis and Haemostasis Subcommittee stated that possible PTS should be detected after 6 months from the index limb deep vein thrombosis, and definitive PTS after 12 months.34 Interestingly, almost 70% of the index VTEs were unprovoked, a known risk factor for PTS in children.35,36 Furthermore, the baseline PTS detected for patients with previous VTE was 17%, which is lower than the weighted mean (43%; 95% CI, 38% to 48%) reported for pediatric PTS detected retrospectively.21 Contemporary work investigated whether DOACs (ie, rivaroxaban) can reduce the rate of PTS in adults with limb deep vein thrombosis in comparison with LMWH/VKA by providing a more stable anticoagulation, but this protective effect was not initially confirmed (odds ratio [OR], 0.76; 95% CI, 0.51-1.13).37 Recently, this suspicion arose again in adults treated with rivaroxaban (OR, 0.5; 95% CI, 0.3-0.9: P = .02).38 Our low PTS rate will need to be reevaluated after a longer follow-up. In our trial, <75% of children experienced adverse events,12% reported serious adverse events, and <6% of children discontinued treatment because of an adverse event. These data are comparable to adult patients treated in the pivotal VTE trials of dabigatran, apixaban and rivaroxaban. In the RE-COVER, RE-MEDY and RE-SONATE trials, 66% to 71% of patients treated with dabigatran experienced adverse events, 7% to 16% reported serious adverse events, and 7% to 9% discontinued treatment because of an adverse event.13,14 Similarly, in the AMPLIFY and EINSTEIN trials, 67% to 71% and 63% of patients treated with apixaban and rivaroxaban, respectively, experienced adverse events; 13% to 16% and 12% reported serious adverse events; and 6% to 8% and 5% discontinued treatment because of an adverse event.39-41
Finally, an analysis of dabigatran pharmacokinetic/pharmacodynamic data from previous pediatric and adult trials indicates that the most appropriate assays for dabigatran might be dTT and ECT,42 which is further supported by data from this trial, as well as the DIVERSITY trial.24 Developmental changes in the hemostatic system had little effect on dabigatran pharmacokinetic/pharmacodynamic relationships, with children aged <2 months being the possible exception.42 A pharmacokinetic simulation analysis of dabigatran exposure in pediatric patients (data on file) has indicated that the impact of dose adjustments on overall exposure is minimal. Therefore, it is considered appropriate to follow an age- and weight-adjusted dosing algorithm in pediatric patients without monitoring of dabigatran plasma levels.
Our trial did not have a comparator arm, yet the present safety data complement the dabigatran safety data from DIVERSITY that does have a comparator arm and showed similar efficacy and safety of dabigatran vs SOC in pediatric patients treated for acute VTE.24 As mentioned previously, the duration of follow-up in our trial from DVT diagnosis to time of PTS assessment during the study was also variable, given the variability in lag time from VTE diagnosis to enrollment in this study of extended VTE treatment.
In view of the low patient numbers and different risk profile of patients in stratum 3 (age, >3 months to <2 years), our data cannot be extrapolated with absolute confidence to younger patients. Based upon the inclusion criteria and the study population (ie, >60% unprovoked VTE), our findings apply most directly to pediatric patients with unprovoked VTE and those who require extended anticoagulant therapy (ie, beyond 3 months post-VTE diagnosis). A final limitation is the exclusion of patients with early withdrawal from the study medication due to failing to reach target dabigatran concentrations not included in the main analyses. However, the results of the sensitivity analyses that included all patients entered, and the full study period (on-treatment plus follow-up), were consistent with those of the primary analyses. Moreover, comparison of baseline patient and thrombus characteristics between patients withdrawn due to failing to reach target dabigatran concentrations vs the retained population shows that there are no statistical differences in the 2 groups (supplemental Table 5).
In conclusion, this trial has shown favorable safety of pediatric formulations of dabigatran for secondary prevention of VTE in children aged from >3 months to <18 years with persistent VTE risk factors.
To ensure independent interpretation of clinical study results, Boehringer Ingelheim grants all external authors access to all relevant material, including participant-level clinical study data, and relevant material as needed by them to fulfill their role and obligations as authors under the International Committee of Medical Journal Editors criteria. Furthermore, clinical study documents (eg, study report, study protocol, statistical analysis plan) and participant clinical study data are available to be shared after publication of the primary manuscript in a peer-reviewed journal and once regulatory activities are complete and other criteria met per the Boehringer Ingelheim Policy on Transparency and Publication of Clinical Study Data: https://trials.boehringer-ingelheim.com/transparency_policy.html. Prior to providing access, documents will be examined, and, if necessary, redacted and the data will be de-identified, to protect the personal data of study participants and personnel, and to respect the boundaries of the informed consent of the study participants. Clinical Study Reports and Related Clinical Documents can be requested via this link: https://trials.boehringer-ingelheim.com/trial_results/clinical_submission_documents.html. All such requests will be governed by a Document Sharing Agreement. Bona fide, qualified scientific and medical researchers may request access to de-identified, analyzable participant clinical study data with corresponding documentation describing the structure and content of the datasets. Upon approval, and governed by a Data Sharing Agreement, data are shared in a secured data-access system for a limited period of 1 year, which may be extended upon request. Researchers should use https://clinicalstudydatarequest.com to request access to study data. Funded by Boehringer Ingelheim; 1160.108, clinicaltrials.gov identifier, NCT02197416.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Sven Wichmann, Carolyn Cook, and Abu Sami for programming support; Alison Monckton for data management support; Joachim Stangier for support with coagulation assays; Dietmar Gansser for support with pharmacokinetic assays; Birgit Kovacs for pharmacovigilance support; and Branislav Biss, Peter Boehm, Lisa Cronin, Axel Dienemann, Ivan Manastirski, and Liljana Pesevski for trial management support. Medical writing assistance, and editorial and technical support in the preparation of the manuscript, was provided by Carolyn Bowler, Natalie Dennis, and Bill Wolvey of Parexel.
This work was supported by Boehringer Ingelheim International GmbH.
Authorship
Contribution: L.R.B., M.A., J.H., L.B., E.C., L.G.M., I.N., P.S., T.K., O.Z., I.T., M.S., F.H., Z.S., J.K., S.G., M.B., and M.L. were responsible for editing the manuscript during development, were involved in the design and execution of the trial, and approved the final draft.
Conflict-of-interest disclosure: L.R.B. is a member of a Pediatric Expert Working Group for Boehringer Ingelheim, and has received Advisory Board fees from Boehringer Ingelheim. M.A. is a member of a Pediatric Expert Working Group for Boehringer Ingelheim, and has received Advisory Board fees from Daiichi Sankyo. J.H. is a member of a Pediatric Expert Working Group for Boehringer Ingelheim and has received honoraria from Boehringer Ingelheim for congress presentation. L.B. is a member of a Pediatric Expert Working Group for Boehringer Ingelheim, and reports fees to her institution from Janssen Pharmaceuticals. E.C. is a member of a Pediatric Expert Working Group for Boehringer Ingelheim, and reports personal fees from Roche, Sobi, Bristol-Myers Squibb, CSL Behring, and Shire/Takeda. L.G.M. is a member of a Pediatric Expert Working Group for Boehringer Ingelheim, reports consultant fees from Pfizer as a steering committee member, and has received a research grant from Bristol-Myers Squibb. P.S. reports personal fees from Takeda and CSL Behring. I.T., M.S., F.H., Z.S., J.K., S.G., and M.B. are all employees of Boehringer Ingelheim. M.L. is a member of a Pediatric Expert Working Group for Boehringer Ingelheim. The remaining authors declare no competing financial interests.
A list of the committees and principal investigators of the DIVERSITY Study Investigators appears in the supplemental appendix.
Correspondence: Matteo Luciani, Hemostasis and Thrombosis Center, Hematology/Oncology Department, Pediatric Hospital Bambino Gesù, Piazza S. Onofrio 4, 00165 Rome, Italy; e-mail: matteo.luciani@opbg.net.
