Key Points
U2 exhibited low rates of immune-mediated toxicities associated with other PI3K-δ, including diarrhea, colitis, pneumonia, and hepatic toxicity.
This combination had promising preliminary activity across a broad range of B-cell malignancies, including a 17% complete response rate.

Abstract
Targeting both CD20 and phosphatidylinositol 3-kinase (PI3K), a protein that is critically involved in B-cell maturation, could be an efficacious strategy for treating B-cell malignancies. The safety of the next-generation compounds umbralisib, a PI3K-δ inhibitor, plus ublituximab, an anti-CD20 monoclonal antibody (combination referred to as U2), was evaluated in patients with chronic lymphocytic lymphoma (CLL) or non-Hodgkin lymphoma (NHL) in this phase 1/1b study. Phase 1 dose escalation was performed with a 3 + 3 design to establish the maximum tolerated dose. In this portion, ublituximab was given intravenously (NHL, 900 mg; CLL, 600 or 900 mg) for 12 cycles. Umbralisib was given orally once daily at 800 or 1200 mg (initial formulation) or 400 to 1200 mg (micronized formulation) in the phase 1 dose escalation portion, and at 800 to 1200 mg in the phase 1b portion until progression, toxicity, or study removal. The maximum tolerated dose was not reached in either the CLL or NHL cohort, and only 1 dose-limiting toxicity was observed. U2 had low instances of grade 3 or higher diarrhea (8%), pneumonia (8%), or hepatic toxicity (4%). Treatment discontinuation due to adverse events occurred in 13% of patients, and umbralisib dose reductions occurred in 15% of patients. The overall response rate for all patients was 46% with 17% complete responses. The median duration of response was 20 months (95% confidence interval, 11.3-not reached). U2 was well tolerated, and no new safety signals were observed over single-agent umbralisib. Preliminary efficacy with this combination is promising and warrants further investigation. This study was registered at www.clinicaltrials.gov as #NCT02006485.
Introduction
Novel drug combinations are poised to broaden the therapeutic options for patients with relapsed or refractory (R/R) B-cell non-Hodgkin lymphoma (B-NHL) and chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL). Anti-CD20 monoclonal antibodies such as rituximab have revolutionized treatment of B-cell malignancies and are a mainstay component of treatment regimens for B-NHL and CLL.1 Phosphatidylinositol 3-kinase (PI3K)-δ inhibitors represent a novel class of drugs with therapeutic potential in B-cell malignancies due to the high and specific expression of PI3K-δ in cells of hematopoietic origin, as well as the upregulation of this protein in lymphoid NHL.2-5 A previous phase 3 trial of idelalisib with rituximab vs rituximab alone showed that the combination resulted in high response rates and promising survival outcomes over single-agent rituximab in R/R CLL6 ; however, idelalisib is associated with considerable toxicities (eg, diarrhea/colitis, infections, hepatic toxicity) that require special management. Newer PI3K inhibitors with activity against the p110-δ isoform, duvelisib and copanlisib, are generally associated with lower rates of hepatic toxicity than idelalisib, but treatment discontinuations due to adverse events (AEs) remain high with these agents.7,8
Ublituximab and umbralisib are novel compounds targeting CD20 and PI3K-δ, respectively, each with promising single-agent activity and safety profiles in CLL and B-NHL. Ublituximab is a next-generation, type 1 chimeric monoclonal antibody targeting a unique epitope on the CD20 antigen that is distinct from rituximab, ofatumumab, and obinutuzumab; it has been glycoengineered to have a low-fucose–containing, fragment crystallizable (Fc) region to allow for enhanced antibody-dependent cellular cytotoxicity.9 Umbralisib is a once-daily PI3K-δ and casein kinase-1ε inhibitor that is structurally distinct from other PI3K-δ inhibitors which are approved, or in development, with improved selectivity for the PI3K-δ isoform.10 The combination of ublituximab and umbralisib has been shown to have synergistic activity in a preclinical study.11
Single-agent studies of ublituximab and umbralisib have reported clinical activity and favorable safety of each of these compounds in CLL and B-NHL. In a phase 1/2 trial, single-agent ublituximab was associated with encouraging response rates and was well tolerated with no dose-limiting toxicities (DLTs) or unexpected adverse events (AEs) identified in patients with R/R CLL and B-NHL. Sustained lymphocyte depletion was reported in patients with CLL, including those refractory to previous rituximab treatment.12 In the phase 1 study of single-agent umbralisib in patients with CLL, B-NHL, or Hodgkin lymphoma, the umbralisib dose was escalated from 50 to 1800 mg once-daily, with the recommended phase 2 dose determined to be 800 mg once daily. Most toxicities were grade 1 or 2 in severity, with the most common events being diarrhea (40% grade 1/2, 3% grade 3), nausea (41% grade 1/2, 1% grade 3), and fatigue (28% grade 1/2, 3% grade 3). Across all doses tested and all disease types, objective responses to umbralisib were seen in 37% of patients with R/R hematological malignancies, and 73% of patients had reductions in disease burden.10
The favorable safety profile of single-agent umbralisib makes it an ideal candidate for combination studies. Single-agent umbralisib has been associated with low rates of grade 3/4 diarrhea and grade 3/4 colitis, occurring in 3% and 2% of patients, respectively, and a long-term safety analysis of umbralisib supports that rates of diarrhea, colitis, transaminitis, and pneumonitis remain low over time.10,13 As a result, various combinations that include umbralisib, such as umbralisib plus ibrutinib, umbralisib plus obinutuzumab and chlorambucil, and ublituximab plus umbralisib plus ibrutinib, are currently under clinical investigation; the studies with interim data available have all resulted in overall response rates (ORRs) of at least 80% in patients with CLL and B-NHL.14-16
Given the critical role of PI3K-δ in B-cell malignancies and the success of anti-CD20/PI3K-δ combination therapy in CLL and B-NHL, targeting CD20 and PI3K-δ with optimized novel agents has the potential to have an improved safety profile over existing doublet therapy while remaining an effective clinical approach.6,17-19 Owing to the encouraging early-phase clinical results with single-agent ublituximab and single-agent umbralisib, and the synergistic activity of the combination in a preclinical study,11 a Phase 1/1b trial of the combination of ublituximab plus umbralisib (combination referred to as U2) was undertaken in patients with R/R B-NHL and CLL/SLL.
Methods
Study design and treatment
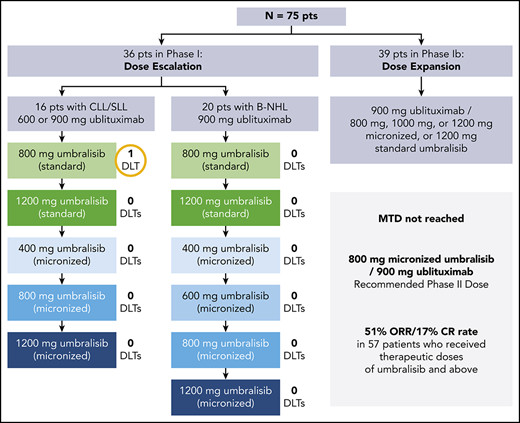
This phase 1/1b multicenter study was conducted at 5 sites in the United States to determine the safety of U2 in R/R B-NHL and CLL. The data cutoff was June 1, 2018. The phase 1 dose escalation portion was performed with fixed doses of ublituximab (900 mg for patients with B-NHL and 600 or 900 mg for patients with CLL) and escalating doses of umbralisib (800 or 1200 mg initial formulation; 400-, 600-, 800-, 1000-, or 1200-mg micronized formulation) to establish the maximum tolerated dose (MTD). Ublituximab dose selection was based on the results of an open-label phase 1/2 trial, which defined the recommended phase 2 dose of ublituximab as 900 mg on the basis of a lack of additional clinical benefit beyond this dose level, and tested doses of 900 mg for patients with NHL, and both 600 and 900 mg for patients with CLL.12 The 600-mg ublituximab dose level was included as the starting dose level for patients with CLL in particular based on a higher rate of infusion-related reactions observed in patients with CLL in the phase 1 study. Umbralisib doses were selected based on a concurrently conducted phase 1 dose escalation trial that assessed umbralisib dose levels of 800 and 1200 mg of the initial formulation and 800, 1000, and 1200 mg of a micronized formulation that showed improved exposure in pharmacokinetic studies, establishing that doses ≥800 mg of the micronized formulation provided umbralisib plasma concentrations, which exceeded the minimum target exposure for the entire 24-hour dosing interval.10 The patients with B-NHL and CLL were accrued and analyzed separately and were treated in cohorts. Dose escalation was performed by using a 3 + 3 design, although intrapatient dose escalation was allowed at the discretion of the investigator after at least 1 cycle of U2 once higher dose cohorts cleared. Select doses not exceeding the MTD were expanded in the phase 1b portion of the study to determine the optimum recommended phase 2 dose. After a time on study of at least 1 year, patients had the option to transfer on to a compassionate use extension study (#NCT03207256).
Patients in the initial cohorts received 12 cycles of intravenous (IV) ublituximab on days 1, 8, and 15 of cycles 1 and 2, and day 1 of cycles 4, 6, 9, and 12. The protocol was amended mid-study to administer ublituximab on days 1, 8, and 15 of cycle 1, day 1 of cycles 2 through 6, and then day 1 of every 3 cycles (cycles 7-12) in alignment with other combination studies using ublituximab.15 All ublituximab infusions were premedicated with a corticosteroid (dexamethasone 10-20 mg or equivalent) and an antihistamine (diphenhydramine 50 mg or equivalent). Dose reductions of ublituximab were not permitted. Patients received oral umbralisib once-daily beginning on day 1 of cycle 1 and continued until progression, toxicity, or study removal.
DLTs were assessed in the first cycle. The DLT criteria for each cohort are listed in the supplemental Appendix (available on the Blood Web site). The MTD was defined as the highest dose at which 0 of 3 patients or 1 of 6 patients experienced a DLT during 1 cycle of therapy. If ≥2 of 6 patients in a cohort experienced a DLT, the MTD was exceeded. After the MTD was established for the B-NHL and CLL cohorts, patients who completed the dose escalation phase were allowed to be dosed at the MTD or re-enter the expansion cohorts (phase 1b) if they had no ongoing toxicity that precluded receiving the dose at the MTD. Additional patients were enrolled to reach a maximum of 60 patients in each of the B-NHL and CLL cohorts. Safety assessments continued on the expanded safety population, defined as all enrolled patients who received at least 1 dose of either drug. All patients were followed up for AEs for 30 days after discontinuation or completion of the treatment, and all new AEs reported during this period were followed up until resolution unless, at the discretion of the investigator, they were not likely to improve because of underlying disease progression.
The protocol was approved by the institutional review board or an ethics committee at each institution, and the study was conducted in accordance with Good Clinical Practice Guideline and the Declaration of Helsinki. All patients provided written informed consent before participation. This trial is registered at www.clinicaltrials.gov (#NCT02006485).
Patient eligibility
Eligible patients were aged ≥18 years with histologically confirmed, measurable, or evaluable B-NHL or CLL as approved by the Medical Monitor and Study Chair. Additional eligibility requirements included an Eastern Cooperative Oncology Group performance status of ≤2 and adequate organ system function, as defined by the following criteria: absolute neutrophil count ≥0.75 × 109/L; platelets ≥50 × 109/L; total bilirubin ≤1.5 times the upper limits of normal (ULN); alanine transaminase (ALT) and aspartate transaminase (AST) ≤2.5 × ULN if no liver involvement or ≤5 × ULN if known liver involvement; and creatinine ≤2.0 mg/dL or calculated creatinine clearance ≥50 mL/min (Cockcroft-Gault method). Patients with B-NHL and CLL were required to be refractory to or to have relapsed after at least 1 previous regimen in the phase 1 component; however, previously untreated patients with CLL were eligible for the phase 1b portion. Patients were considered refractory to immediately prior therapy or CD20-directed therapy if they progressed during or within 6 months of treatment. Patients were ineligible if they were currently receiving cancer therapy or any investigational drug within 21 days of treatment initiation, receiving limited palliative radiation within 14 days of treatment initiation, or had received an autologous hematologic stem cell transplant or an allogeneic hematologic stem cell transplant ≤3 months and ≤12 months before study entry, respectively.
Study objectives
The primary study objectives were to assess the safety of the U2 combination, determine the incidence of DLTs, and establish the MTD. All AEs were evaluated according to National Cancer Institute Common Terminology Criteria for Adverse Events v4.0, and CLL hematologic toxicity was graded according to the International Workshop on Chronic Lymphocytic Leukemia guidelines.20 The secondary objectives were to determine the ORR (defined as the sum of complete response [CR] and partial response [PR]) and duration of response (DOR). Response was assessed according to the Revised Response Criteria for Malignant Lymphoma for B-NHL and the 2008 International Working Group CLL Working Guidelines for CLL.20,21 Computed tomography imaging was performed at screening, and patient response was assessed by using computed tomography imaging at ∼8 weeks from initiation of U2 and every 12 weeks thereafter through month 12. After month 12, efficacy assessments were to occur at least every 6 months or at the treating physician’s discretion.
Statistical analysis
Primary efficacy analyses were performed on all enrolled patients who had at least 1 posttreatment efficacy measurement. Early termination of the trial was recommended if the response rate was ≤30% (ie, ≤6 responders) among the first 20 enrolled patients in either cohort. The response rate was assessed by a 90% one-sided confidence interval (CI). DOR was summarized by using descriptive statistics (mean, standard deviation, and median).
Results
Patients and disposition
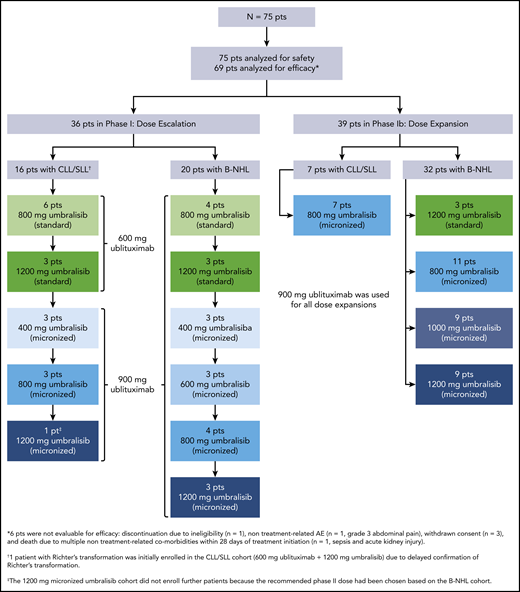
From February 2014 to March 2017, a total of 75 patients were enrolled in the study and received treatment according to the schema in Figure 1; all 75 patients were evaluable for safety, and 69 patients were evaluable for efficacy. Twenty-two patients enrolled had CLL/SLL, and 53 patients enrolled had other forms of B-NHL, including Richter’s transformation, diffuse large B-cell lymphoma (DLBCL), mantle cell lymphoma, follicular lymphoma (FL), and marginal zone lymphoma (MZL) (Table 1). Collectively, patients had a median age of 64 years (range, 26-86 years). The patient population received a median of 3 (range, 0-10) previous therapies and a median of 2 (range, 0-7) previous CD20-directed monoclonal antibody-based regimens. Nine patients had previous Bruton tyrosine kinase (BTK) inhibitor therapy. Fifty-seven percent of patients were refractory to the immediately prior treatment, and 54% were refractory to CD20-directed therapies. Of the 22 patients with CLL/SLL, 9 (41%) had 17p deletion and 6 had previous BTK inhibitor therapy. Cell of origin subtype, as determined according to immunohistochemistry by a local laboratory,22 was available for 20 of 26 patients with DLBCL. The majority of patients with DLBCL (16 of 26 [62%]) had the germinal center B cell (GCB) subtype, and 4 (15%) of 26 had a non-GCB subtype.
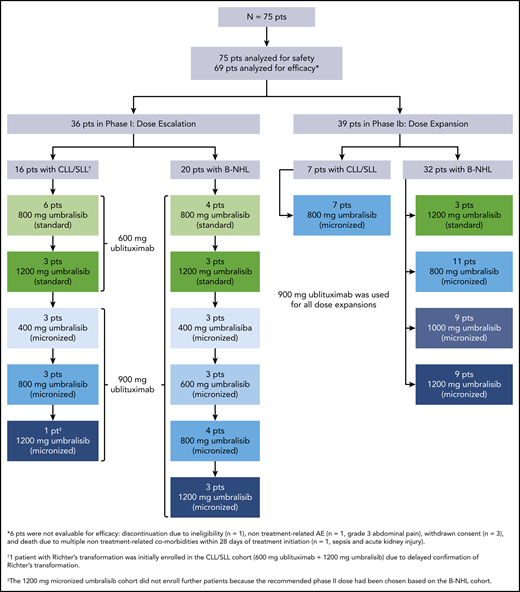
As of the data cutoff, 67 (89%) of 75 patients have discontinued treatment, primarily due to disease progression (44 of 75 [59%]) or AEs (10 of 75 [13%]). Three patients (4%) discontinued treatment because they attained a CR and pursued stem cell transplant, 7 (9%) withdrew consent, and 3 (4%) discontinued for other reasons. Eight patients (11%) remain on study and have a median follow-up of 39 months (Table 2). The median treatment duration was 7.4 months (range, 0.3-52.7 months) for all 75 patients, 11 months (range, 2-40 months) for patients with CLL/SLL, 10 months (range, 1-44 months) for patients with indolent B-NHL, and 2 months (range, 0.3-52.7 months) for patients with aggressive B-NHL. The median treatment duration for patients who received the recommended phase 2 dose of 900-mg ublituximab and 800-mg micronized umbralisib was 9.9 months (range, 0.6-52.7 months).
Dose reductions of umbralisib occurred in 11 patients (15%). Twenty-nine patients (39%) had a temporary hold of umbralisib (most common reasons included grade 3/4 neutropenia [n = 13], grade 3/4 diarrhea [n = 4], and grade 3/4 nausea [n = 2]), and 31 (41%) had a temporary hold of ublituximab (most common reason was infusion-related reaction, of which only 1 patient had grade 3 severity).
Maximum tolerated dose
One DLT was reported in a patient with CLL (treated with 600-mg ublituximab and 800-mg initial formula umbralisib) who had baseline grade 3 neutropenia at study entry that worsened to grade 4, requiring enrollment of an additional 3 patients at this dose level. No additional DLTs were observed during the study, and the MTD was not reached. There were no DLTs in the B-NHL cohorts, and, similar to the CLL cohort, the MTD was not reached. Expansion cohorts received umbralisib at a micronized dose of 800, 1000, or 1200 mg or a standard dose of 1200 mg in the phase 1b portion. Doses of 900-mg ublituximab and 800-mg daily umbralisib were chosen for the combination for ongoing trials in B-NHL and CLL patients due to a lack of additional clinical benefit at higher doses.10,12 Of the patients treated with umbralisib 800 mg, 83% received all intended doses of U2 in the first 2 cycles of therapy.
Safety
The median follow-up for the safety population was 7.4 months (range, 0.3-52.7 months). The majority of AEs in the total population were grade 1 or 2 in severity (Table 3). The most common AEs of any grade were diarrhea, nausea, and fatigue. Any-grade diarrhea was less prevalent in patients with aggressive B-NHL histologies (48%) than in those with CLL/SLL (63%) or indolent B-NHL (71%). Diarrhea had a median time to onset of 21 days (range, 1-838 days) and resolved in a median of 7 days (range, 1-191 days). Grade 3/4 diarrhea was reported in 6 patients (8%); 5 cases were treated with a dose hold, 3 cases were treated with an antidiarrheal, and all cases resolved. Four of the 6 cases of grade 3/4 diarrhea occurred in patients with indolent B-NHL. There was 1 case of biopsy-confirmed colitis, which was treated with treatment hold and steroids. Any-grade neutropenia was reported in 24 patients (32%), with a median time to onset of 49 days (cycle 2) and a median time to resolution of 7 days (range, 1-136 days). Grade 3/4 neutropenia was reported in 21 patients (28%) in the total population and was more prevalent in patients with CLL/SLL (occurring in 50%) than in those with aggressive B-NHL (21%) or indolent B-NHL (17%). Other grade 3/4 events observed in >5% of patients in the total population included pneumonia (n = 6 [8%]) and abdominal pain (n = 5 [7%]). Grade 3/4 increases in ALT and AST levels occurred in 2 patients (3%) and 1 patient (1%), respectively, all of whom had indolent B-NHL. One of the patients with transaminitis underwent steroid treatment, and 1 case resolved without intervention. The median time to onset for ALT/AST elevations of any grade was 55 days. There were 2 cases of pneumonitis, which resolved in a median of 21 days (range, 15-28 days). One patient died while on the study due to septic shock, respiratory failure, and acute kidney injury, all of which were determined not to be related to either study drug. Three patients experienced grade 3/4 nausea; all resolved without treatment.
Efficacy
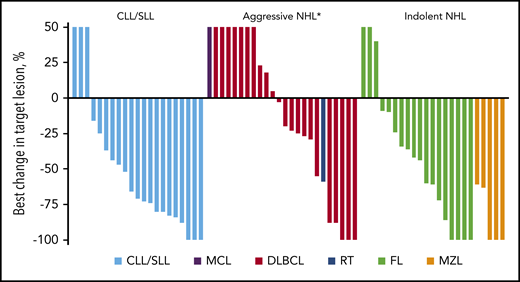
Sixty-nine patients treated with any dose of umbralisib were eligible for efficacy analysis, and the median follow-up time in this population was 8 months (range, 0.6-52.7 months); 32 (46%) achieved an overall response (CR + PR), including 12 (17%) with a best response of CR. The median time to first response was 8 weeks (range, 8-44 weeks), and the median DOR was 20 months (95% CI, 11.3-not reached [NR]). The median DOR for patients in CR was 35.3 months (range, 5.6-48.3 months). Best responses according to histology are shown in Table 4 (supplemental Figure 2 presents DOR curves according to histology). The ORR was 100% in MZL, 62% in CLL, and 44% in FL. The ORR in DLBCL was 23%, with responses in 3 of 12 patients with the GCB subtype and in 1 of 4 patients with the non-GCB subtype. Patients with MZL had the longest DOR, with a median NR (95% CI, 3.1-NR); those with DLBCL had the shortest DOR, with a median of 16 months (95% CI, 5.6-NR). The depth of responses across different histologies is shown in Figure 2.

Best percent change from baseline in disease burden. The best percent change in tumor size for 68 patients eligible for efficacy analysis was determined by the principal investigators. One patient was not evaluable for change in tumor size due to lack of a post-baseline scan. Each bar represents an individual patient. Patients are grouped according to histology. MCL, mantle cell lymphoma; RT, Richter’s transformation. *Post-baseline scan was not available for 1 patient with MCL.
Best percent change from baseline in disease burden. The best percent change in tumor size for 68 patients eligible for efficacy analysis was determined by the principal investigators. One patient was not evaluable for change in tumor size due to lack of a post-baseline scan. Each bar represents an individual patient. Patients are grouped according to histology. MCL, mantle cell lymphoma; RT, Richter’s transformation. *Post-baseline scan was not available for 1 patient with MCL.
Efficacy was further examined in the subset of patients who received umbralisib at what was considered to be therapeutic-dose levels as determined in the phase 1 first-in-human study.10 This therapeutic-dose subset included patients receiving at least 1200 mg of non-micronized or ≥600 mg of micronized umbralisib, including the recommended phase 2 dose of 800 mg. Of the 57 patients treated with therapeutic-dose umbralisib, 29 (51%) had a response, with 12 (21%) having CRs after a median time on drug of 9.9 months (range, 0.6-52.7 months) (Table 5). Virtually all responding patients (29 of 32) had received therapeutic-dose levels of umbralisib. The median DOR in this subset of patients was NR (95% CI, 13.65-not estimable) (supplemental Figure 1). Response rates were elevated slightly in this group of patients, particularly in those with CLL/SLL (ORR, 67%) and those with FL (ORR, 53%) compared with the total population, albeit with small numbers of patients. The response rate among those patients with CLL/SLL and previous BTK therapy was 40% (2 of 5). The rate of response did not seem to differ among patients with NHL treated at 800, 1000, or 1200 mg of micronized umbralisib; therefore, ublituximab 900 mg in combination with umbralisib 800 mg was determined to be the recommended phase 2 dose. The median DOR for all patients with CLL/SLL was 25.89 months (95% CI, 5.03-not estimable) (supplemental Figure 2). The median progression-free survival in this group of patients was 15.7 months (95% CI, 7.37-NR), with progression-free survival according to histology of 27.57 months (95% CI, 4.77-not estimable) for CLL/SLL, 7.37 months (95% CI, 1.38-12.93) for aggressive B-NHL, and NR (95% CI, 5.76-not estimable) for indolent B-NHL (supplemental Figure 3).
Discussion
As novel agents have emerged for patients with B-cell malignancies, there is a need for rational approaches in designing nonchemotherapy-based combinations, with the selection of well-tolerated and combinable agents of particular importance. Ublituximab and umbralisib as individual agents have manageable toxicity profiles with activity in CLL and R/R B-NHL.10,12 In the current study, the U2 combination had no cumulative toxicity signal that could be considered as overlapping, deleterious, or unexpected compared with the AE profile of either agent alone at similar doses,15,23 with no DLTs in the B-NHL cohort and 1 DLT in a neutropenic CLL patient without allowance of growth factor use in cycle 1. Notably, the MTD was not reached in either the CLL or the B-NHL cohort. Dose-dependent activity was evident at 600 mg of micronized umbralisib and higher without a significant exacerbation of the AE profile in the total patient population. The final recommended doses for the CLL and B-NHL cohorts were 900 mg IV of ublituximab and 800 mg of oral umbralisib daily. The dose intensity during cycles 1 and 2 was acceptable with respect to AEs, with 83% of patients treated at 800 mg of umbralisib receiving all intended doses of U2 in the first 2 cycles of therapy.
Discontinuations of U2 due to AEs were limited (13%). The safety profile of U2 differed slightly between histologies, with the incidence of any-grade transaminitis, diarrhea, and nausea being lower in patients with aggressive NHL, possibly owing to a reduced treatment time in these patients. Overall, the majority of drug-related AEs were grade 1 or 2, and the incidence of severe or life-threatening AEs that were attributed to the U2 combination were low after a median time on study of 7.4 months. For the notable AEs of diarrhea, neutropenia, and pneumonitis, the time to resolution was generally short, within a median of 7 days for diarrhea and neutropenia and a median of 21 days for pneumonitis. The only grade 3/4 AE reported in >10% of patients was neutropenia, which was appropriately supported by granulocyte-colony stimulating factor to maintain dose intensity. The median time to onset of neutropenia was relatively early, occurring in cycle 2, and was greater than that observed with umbralisib monotherapy in its first-in-human phase 1 study, suggesting an association with ublituximab administration.10 Colitis and hepatic toxicity were nearly absent in patients treated with the U2 combination with this length of follow-up, which are consistent with findings from a recent study of single-agent umbralisib that showed grade 3 or higher increases in AST and ALT levels observed in 3% of patients; grade 3 or higher colitis was observed in 2% of patients. Results from the phase 3 DUO (Duvelisib vs Ofatumumab in Relapsed/Refractory CLL/SLL) trial of single-agent duvelisib in patients with R/R CLL reported grade 3 or higher colitis in 12% and severe opportunistic infections in 6% of patients after a median treatment time of ∼11.5 months (50 weeks), which is notably longer than the median duration of treatment of 7.4 months in the current study.8 The incidence of opportunistic infections was also relatively low with U2 treatment, with no events of Pneumocystis jirovecii pneumonia or cytomegalovirus infection observed, albeit with shorter follow-up and a heterogeneous patient population. Anti-infectious prophylaxis was not required on study but was encouraged at investigator discretion; overall, 17% of patients were receiving some type of anti-infectious prophylaxis. The results of this study support findings from a long-term integrated analysis of umbralisib safety that reported grade 3 diarrhea in 8% with colitis in only 1 patient, grade 3 or higher transaminitis in 3%, and pneumonitis in 2% of patients after a median duration of exposure of 1.3 years.13 These findings are notable in light of the termination of 7 idelalisib clinical trials because of an increased risk of mortality from infections in patients treated with idelalisib combinations, which has necessitated a Risk Evaluation and Mitigation Strategy to manage toxicity.24
The U2 combination exhibited promising preliminary activity across a broad spectrum of B-NHL/CLL malignancies with an ORR of 46%, warranting further assessment as a combination. These findings are particularly noteworthy when considering that more than one-half of the patient population was refractory to previous CD20 therapy. The CR rate of 17% in this population of R/R patients compares favorably with the CR rate of 8% observed in a previous study of duvelisib plus rituximab and/or bendamustine in patients with relapsed CLL or B-NHL.25 In addition, there was a compelling 100% ORR in patients with MZL (3 CR and 2 PR), with a median duration of response that was NR and the longest responder now at 40 months. Most responses to U2 were observed in the group of patients who received a higher dose of umbralisib, including the recommended phase 2 dose. The safety profile seen collectively in the expansion cohort shows the potential ability to pair the U2 regimen with standard chemotherapy or other novel B-cell–targeting agents that could elevate ORR and prolong DOR in both CLL and appropriate B-NHL subtypes.
The U2 combination was generally well tolerated with relatively low rates of immune-mediated toxicities and opportunistic infections. The daily oral dosing of umbralisib combined with ublituximab allowed optimal dose intensity in a population that included refractory patients with CLL/SLL and B-NHL. Longer follow-up is necessary to fully understand the incidence of potential late-onset toxicities. The limitations of this phase 1/1b study include the lack of a requirement of subtype assessment in DLBCL and a lack of correlative studies to analyze the mechanisms of resistance to dual antibody/PI3K-δ therapy. Additional limitations include the lack of assessment of some prognostic factors for CLL (eg, complex karyotype), the limited sample sizes within specific histologies and the absence of pharmacodynamic/pharmacokinetic assessment of U2. The preliminary compelling activity of the U2 combination observed in multiple subtypes of B-NHL and CLL has informed future studies of this regimen, including the registration-directed phase 3 UNITY-CLL (Ublituximab + Umbralisib Compared to Obinutuzumab + Chlorambucil in Patients With Untreated and Previously Treated Chronic Lymphocytic Leukemia; NCT02612311) and the phase 2/3 randomized UNITY-NHL (Study to Assess the Efficacy and Safety of Ublituximab + Umbralisib With or Without Bendamustine and Umbralisib Alone in Patients With Previously Treated Non-Hodgkin’s Lymphoma; NCT02793583) studies that are currently open globally. In addition, these data have informed ongoing studies using U2 as a backbone therapy for further combination trials, including a study of U2 plus pembrolizumab in CLL and Richter’s transformation (NCT02535286), a study of U2 plus bendamustine in NHL (NCT02793583), and a study of U2 plus ibrutinib in CLL and NHL (NCT02006485).
These data were previously presented at the 2015 American Society of Hematology Annual Meeting, Orlando, FL, 5-8 December 2015.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the patients and caregivers for their participation in this study. They extend further thanks to Michael Chen of TCM Groups, Inc., for assistance with statistical analyses. Medical writing support was provided by Allison Cherry of Nexus Global Group Science, LLC, and funded by TG Therapeutics.
Authorship
Contribution: M.L., P.S., H.P.M., M.S.W., and S.O. designed the analysis and interpreted the data; J.V., L.N., N.F., J.A.B., W.G.W., M.T.S., T.S., C.R.F., and J.B.C. collected data; M.L., P.S., and H.P.M. provided input on the analysis and data interpretation; M.L., P.S., and H.P.M. performed statistical analyses and interpreted the data; M.L., P.S., and H.P.M. drafted the manuscript; and all authors had access to the study data, critically reviewed or edited the manuscript, and approved the final version of the manuscript for submission.
Conflict-of-interest disclosure: M.L. has received grants from Celgene, Curis, Janssen Scientific Affairs LLC, Juno Therapeutics, Pharmacyclics, and TG Therapeutics; and had a consultancy role with AbbVie, ADC Therapeutics, AstraZeneca/Acerta, Bayer, Celgene, Genentech, Gilead Sciences Inc., Janssen/Pharmacyclics, Juno Therapeutics, Kite, Portola, Sanofi-Genzyme, Seattle Genetics, Spectrum, TG Therapeutics, and Verastem. J.V. had a consultancy role with AbbVie, Novartis, Epizyme, Roche, Legend Pharmaceuticals, Kyopharm, Sandoz, Janssen/Pharmacyclics, Kite, Acerta, Nordic Nanovector, and AstraZeneca. L.N. has received personal fees, research support, and honorarium from TG Therapeutics and Celgene, and personal fees and honorarium from Gilead, Novartis, and Spectrum Pharmaceuticals. N.F. had an advisory role with Celgene, Roche, and Janssen; and received grants from AbbVie. J.A.B. has received grants from Pharmacyclics and Gilead and honoraria from Janssen. T.S. has received grants and nonfinancial support from TG Therapeutics. C.R.F. had a paid consultancy role with Spectrum, Celgene, Denovo Biopharma, Seattle Genetics, Gilead, Bayer, Karyopharm, AstraZeneca, and Beigene; an unpaid consultancy role with Genentech/Biogen Idec/Roche and Millennium/Takeda; received research funding from AbbVie, Acerta, Celgene, Gilead, Infinity Pharmaceuticals, Janssen, Millennium/Takeda, Spectrum, Onyx Pharmaceuticals, and Pharmacyclics; and received payments for educational materials from Clinical Care Options, Educational Concepts, PRIME Oncology, and Research to Practice. J.B.C. has received grants from Seattle Genetics, Takeda, Novartis, American Society of Hematology, and Lymphoma Research Foundation; and received personal fees from Seattle Genetics and Genentech. S.O. had a consultancy role with Amgen, Astellas, Celgene, GlaxoSmithKline, Janssen Oncology, Aptose Biosciences, Inc., Vaniam Group, LLC, AbbVie, Alexion Pharmaceuticals, Gilead, Pharmacyclics LLC, an AbbVie Company, TG Therapeutics, Pfizer, and Sunesis; and has received research support from Kite, Regeneron, Acerta, Gilead, Pharmacyclics LLC, an AbbVie Company, TG Therapeutics, Pfizer, and Sunesis. P.S., H.P.M., and M.S.W. report employment and equity ownership with TG Therapeutics. The remaining authors declare no competing financial interests.
Correspondence: Matthew Lunning, Division of Oncology & Hematology, University of Nebraska Medical Center, 986840 Nebraska Medical Center, Omaha, NE 68198; e-mail: mlunning@unmc.edu.