


Abstract
HLA-haploidentical hematopoietic stem cell transplantation is now one of the most commonly employed alternative donor techniques, with most centers applying T-cell–replete strategies such as that developed by the Baltimore group using high-dose posttransplant cyclophosphamide. HLA-haploidentical hematopoietic stem cell transplantation using posttransplant cyclophosphamide is associated with low rates of severe graft-versus-host disease and nonrelapse mortality and does not require graft manipulation or storage, which results in a low graft acquisition cost. Its remarkable safety when used with reduced-intensity conditioning has been demonstrated in patients up to 75 years old with outcomes similar to those of patients in their 50s. Several large, registry-based retrospective studies have confirmed the efficacy of HLA-haploidentical hematopoietic stem cell transplantation with posttransplant cyclophosphamide, achieving results comparable to those of HLA-matched hematopoietic stem cell transplantation. In this article, we describe our approach to this rapidly available and clinically simple platform and address some of the key clinical questions associated with its use.
Introduction
Over the past two and one-half decades, several methods of facilitating HLA-haploidentical hematopoietic stem cell transplantation (haplo-HSCT) have been developed. The 3 most frequently used platforms consist of (1) posttransplant cyclophosphamide (PTCy); (2) granulocyte colony-stimulating factor priming, intensive postgrafting immunosuppression, and antithymocyte globulin using combined peripheral blood stem cell and bone marrow (BM) allografts; and (3) T-cell depletion with either “megadose” CD34+ cells or selected α/β T-cell and B-cell depletion. Owing in part to the development of T-cell–replete strategies such as the use of PTCy,1 which has been associated with survival outcomes comparable to those of HLA-matched HSCT,2-6 there has been a rapid expansion in haplo-HSCT. For instance, among centers within the European Society for Blood and Marrow Transplantation, the use of haploidentical donors grew by 291% from 2005 to 2015.7 A previous review summarized the most often applied T-cell–replete and T-cell–depleted haplo-HSCT strategies,8 but the present article focuses on PTCy-based haplo-HSCT, which is cost-effective, safe, and easily replicated, leading to its widespread use.
An advantage of haplo-HSCT is the rapidity and near-universality of identifying haploidentical donors and their availability for subsequent stem cell or lymphocyte donation to treat relapse or boost engraftment. For instance, at Johns Hopkins Hospital from 2006 to 2011, 96.6% of recipients had one or more haploidentical donors, with an average of 2.5 donors per candidate. Given that multiple donors are often available for a given recipient, determining which donor factors influence outcomes is particularly important in haplo-HSCT. Several studies have been published in the past few years to try to improve donor selection alogrithms.9-13 One of the most important facets of donor selection that is unique to HLA-mismatched transplantation are donor-specific antibodies (DSA), which, if present, can require additional desensitization techniques to mitigate the risk of graft failure or may even preclude the use of a given donor.14,15
Given its increased use, questions that have previously been addressed in HLA-matched HSCT are now being asked in haplo-HSCT. For instance, the merits of increasing conditioning intensity, differing graft sources, and disease status are being examined in haplo-HSCT using PTCy, with conclusions mostly akin to those in HLA-matched HSCT. Although survival outcomes, including relapse, after haplo-HSCT and HLA-matched HSCT appear to occur at similar rates, it is important to recognize that a distinct immune mechanism with significant clinical implications, called “HLA loss,” occurs in 30% of acute myeloid leukemia (AML) relapses after haplo-HSCT.16-18 The present article discusses a complicated case of one patient with AML to review what we have learned in the last decade of research and describes the authors’ personal approach to haplo-HSCT with PTCy.
Clinical case
A 58-year-old African American man with insulin-dependent diabetes, hypertension, and depression controlled with a selective serotonin reuptake inhibitor was diagnosed with AML with a complex karyotype after presenting with leukocytosis, anemia, and thrombocytopenia. He underwent initial management with fluids, hydroxyurea, and allopurinol, then initiation of induction chemotherapy with cytarabine and daunorubicin (7 + 3). His clinical course was complicated by neutropenic fever that resolved with cefepime. A day 14 BM biopsy showed no residual AML. On his recovery bone marrow biopsy, he achieved a morphologic complete remission (CR), but with residual cytogenetic abnormalities. At the time of count recovery, he was referred for discussion of HSCT. His HSCT-specific comorbidity index score was 2. The patient’s immediate family included an 88-year-old father, an 85-year-old mother, 2 brothers aged 60 and 52 years old, and 1 sister who was 65 years old. He did not have any biological children. Both parents shared one HLA haplotype with the patient; the younger brother and his sister also shared one HLA haplotype; and the older brother was disparate (Figure 1). Class I and class II screens for DSA were negative. A preliminary unrelated donor search showed 3 potential donors who had a low probability of being a 10/10 HLA match.
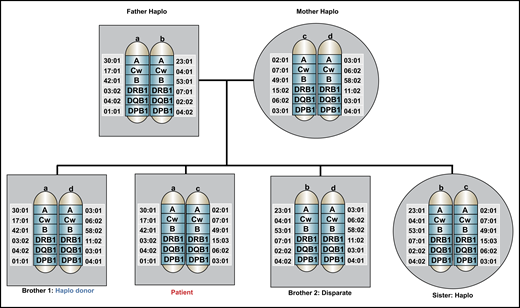
Family pedigree of the patient in the clinical case. An HLA-haploidentical donor has inherited one HLA haplotype in common with the recipient and is mismatched for anywhere between 0 and 5 HLA genes on the unshared haplotype. Biological parents and biological children always share an HLA haplotype with the recipient, unless a rare genetic rearrangement has occurred. In this pedigree analysis, the patient has 3 siblings, none of whom are HLA-matched, but 2 are partially matched related (haploidentical) donors, whereas 1 brother is disparate. Other potential HLA-haploidentical donors include half-siblings, aunts, uncles, nieces, nephews, cousins, or grandchildren.
Family pedigree of the patient in the clinical case. An HLA-haploidentical donor has inherited one HLA haplotype in common with the recipient and is mismatched for anywhere between 0 and 5 HLA genes on the unshared haplotype. Biological parents and biological children always share an HLA haplotype with the recipient, unless a rare genetic rearrangement has occurred. In this pedigree analysis, the patient has 3 siblings, none of whom are HLA-matched, but 2 are partially matched related (haploidentical) donors, whereas 1 brother is disparate. Other potential HLA-haploidentical donors include half-siblings, aunts, uncles, nieces, nephews, cousins, or grandchildren.
Questions
Is there benefit in waiting for a completed matched unrelated donor search vs proceeding directly with a haploidentical donor?
Identifying fully HLA-matched unrelated donors (MUDs) is difficult for some ethnic groups, reflecting both their HLA diversity and underrepresentation in donor registries. There is also a sense of urgency for patients with high-risk acute leukemia, which may preclude the time required to identify an unrelated donor in the registry. Over recent years, haploidentical donors have increasingly been adopted as a valid, immediately available donor source when an HLA-matched sibling donor (MSD) is unavailable. However, there are certain clinical scenarios in which a MUD or an HLA-mismatched unrelated donor (MMUD) is preferred. These include recipients with familial genetic syndromes; very high levels of DSA to family members; or those who lack living relatives, are estranged from their families, or are adopted and have no children.
In the absence of a definitive indication for an unrelated donor search, is waiting for an unrelated donor beneficial? Data are growing on the similarity in outcomes between haplo-HSCT and both MUD-HSCT and MSD-HSCT. In one of the earliest studies comparing HLA-matched HSCT with haploidentical bone marrow transplantation (haplo-BMT) employing PTCy, we used the Disease Risk Index to show risk-stratified outcomes. In that study, 3-year overall survival (OS) rates were 70% and 73% in low-risk disease, 47% and 49% in intermediate-risk disease, and 25% and 37% in high/vs high-risk disease in HLA-matched and haplo-HSCT, respectively.2 Later that year, in a large Center for International Blood and Marrow Transplant Research (CIBMTR) analysis, Ciurea et al showed that, among patients with AML in complete remission (CR), there was no significant difference in 3-year probability of OS after MUD with calcineurin inhibitor–based prophylaxis and haplo-HSCT with PTCy with either myeloablative conditioning (MAC) or reduced-intensity conditioning (RIC).3 In another CIBMTR study, there were no significant differences in grades II to IV acute graft-versus-host disease (aGVHD), relapse, nonrelapse mortality (NRM), or OS for AML in CR1 after MSD- and haplo-HSCT, but there was less chronic graft-versus-host disease (cGVHD) in recipients of haploidentical grafts, regardless of conditioning or graft source, owing to the use of PTCy.19 For poor-risk AML in CR, when all graft sources were compared, MSD-, MUD-, and haplo-HSCT did not significantly differ with regard to OS, but cord blood transplantation and 9/10 MUD were associated with inferior survival.20
In an analysis of patients with AML aged 60 years and older, haplo-HSCT with PTCy and MUD-HSCT were not associated with significant differences in OS and leukemia-free survival (LFS), but MUD-HSCT was associated with a higher incidence of extensive cGVHD.21 In contrast, in 2 studies in older recipients, survival when using MUD donors younger than age 4022 or using an MSD23 was improved when compared with haploidentical donors, owing to less relapse.22 The authors concluded that this difference may be due to a greater use of peripheral blood stem cell transplantation (PBSCT) in recipients of both MSD-HSCT23 and MUD-HSCT22 in those studies, which has been associated with less relapse24,25 and improved survival25 in these platforms. In a single-center study comparing haplo-PBSCT with PTCy with MUD-PBSCT, there were no significant differences in survival outcomes.26
Given the many studies showing equivalency between HLA-matched HSCT and haplo-HSCT,5 our practice is to not routinely formalize a MUD search unless contraindications to haplo-HSCT exist or no eligible physically and psychologically fit haploidentical donors are identified. However, a currently enrolling Bone Marrow Transplant Clinical Trials Network (BMT CTN 1702) study is asking whether the strength of the preliminary MUD search can be used to decide whether awaiting a MUD or proceeding with an alternative donor results in better outcomes.
Donor selection: do donor relationship, cytomegalovirus serostatus, and age matter?
One of the unique aspects of haplo-HSCT with PTCy is that there is no need to minimize the extent of mismatch between donor and recipient.27 Although we do not currently use the number of mismatches to choose between haploidentical donors, this may change as our understanding of the immune effects of each individual HLA allele expands. For instance, certain preliminary studies have demonstrated improved progression-free survival with class II mismatching in HLA-DRB1 and HLA-DPB1.13,28,29 In the absence of a negative effect of increasing HLA mismatch on outcomes,9,12,13 other donor selection criteria have gained more attention.11 A traditional risk factor for poor outcomes with HLA-matched HSCT has been mismatched donor–recipient cytomegalovirus (CMV) serostatus. However, 2 haplo-HSCT studies showed that donor CMV serostatus was not associated with outcomes.30,31 This may be due to the high rate of CMV reactivation in all CMV-positive recipients undergoing haplo-HSCT, which has led to interest in escalating antiviral prophylaxis to prevent CMV in this population32 as well as the need to investigate more active agents such as letermovir.33
Given the data showing that younger donor age improved outcomes in recipients of MUD allografts,34-36 donor age is of keen interest in haplo-HSCT, where multiple donors are often available. Understanding the impact of donor age on outcomes in haplo-HSCT is difficult, not only because of the correlation between donor and recipient age, but also because of the correlation of recipient age and donor relationship inherent to haploidentical related donors; that is, older recipients are more likely to have children or sibling donors, whereas younger recipients are more likely to have sibling or parent donors. For instance, in a CIBMTR analysis, mortality risk was higher when donors were 30 years or older; however, when patient age, which negatively impacts outcomes, was entered into the model, the effect of donor age was negated.10 In that study, although donor age did not affect outcomes, parent donors were associated with more graft failure (14% compared with 6% to 7% with siblings and offspring), but this had no effect on survival.10 In a subsequent study by the European Society for Blood and Marrow Transplantation in patients with acute leukemia over the age of 40 years, donors over 40 years old were associated with higher NRM and inferior OS and LFS.37 However, in recipients under 40 years old, donor age did not influence outcomes.37 Recently, we have demonstrated the safety of second-degree relative haplo-HSCT with PTCy using predominantly nephews, nieces, and grandchildren.38 These data raise the following yet unanswered questions:
In older recipients without children, should we use younger second-degree donors (nieces or nephews) rather than haploidentical sibling donors?
For recipients with children over 40 years old, should we use grandchildren rather than offspring donors?
Does conditioning intensity matter?
The vast majority of data demonstrating the efficacy of haplo-HSCT with PTCy is associated with the widely accepted RIC approach that consists of fludarabine/cyclophosphamide and low-dose total body irradiation (TBI), with recent interest in increasing the intensity of TBI from 200 cGy to 400 cGy to further reduce relapse and graft failure. However, far less consensus exists with regard to the preferred MAC regimen for haplo-HSCT with PTCy. This is in part due to a wide range of conditioning platforms used, including busulfan/cyclophosphamide, fludarabine/TBI, fludarabine/busulfan and thiotepa, fludarabine/melphalan and thiotepa, or fludarabine/busulfan, each of which has been studied in relatively small patient populations with encouraging results. For instance, fludarabine/busulfan and thiotepa haplo-BMT with PTCy39 and fludarabine/TBI haplo-PBSCT with PTCy40 were associated with a cumulative incidence of relapse of 24% at 4 years39 and 2 years,40 respectively, with low associated NRMs.
A recurring question in HSCT is whether the intensity of the preparative regimen impacts outcomes. In haplo-HSCT, several studies have sought to examine this question. For instance, when we compared PTCy platforms including MAC MSD, MAC MUD, and RIC haplo-HSCT, we found that composite GVHD-free and relapse-free survival endpoints were not significantly different.6 Similarly to HLA-matched HSCT, there was less relapse with MAC but more GVHD and NRM, leading to comparable composite outcomes. However, this analysis compared intensity in differing donor sources, and the haploidentical cohort received mycophenolate mofetil (MMF) and tacrolimus as GVHD prophylaxis in addition to PTCy. In a propensity score–adjusted analysis of only haplo-HSCT, when compared with MAC HSCT, RIC HSCT was associated with no significant differences in OS or disease-free survival, but it was associated with higher relapse and less NRM.41 Rates of aGVHD and cGVHD were not different by intensity of conditioning.41 In patients over 60 years old, Santoro et al showed no significant difference in NRM, relapse, OS, or LFS in patients treated with MAC or RIC haplo-HSCT with PTCy.21 Although data comparing MAC vs RIC exclusively in younger patients does not exist, we typically reserve MAC conditioning for very fit younger candidates in whom we anticipate the risk of NRM will be exceedingly low.
Does graft source matter?
In the original studies of haploidentical donor transplantation, BM was the graft source used.1,42 However, some centers prefer PBSCT, which does not require anesthesia and also avoids the logistics of obtaining operating room time and harvesters. Given that PBSCT has been associated with less graft failure25,43 and quicker engraftment in multiple studies of HLA-matched HSCT,44-48 there has been clinical interest in using PBSCT for diseases that are associated with difficult engraftment, such as myeloproliferative neoplasms and myelodysplastic syndrome. In haplo-HSCT with PTCy, neutrophil engraftment is, on average, 1 to 2 days earlier with PBSCT, but rates of graft failure seem to be similar between the sources.24,49,50 However, a comparison of graft sources for myeloproliferative neoplasms and myelodysplastic syndrome has not been performed.
In one of the earliest studies to compare graft sources after RIC haplo-HSCT with PTCy, Castagna et al demonstrated in 69 patients that there were no significant differences in survival outcomes.49 In 2017, a CIBMTR study including a variety of hematologic malignancies enriched for RIC cases found that although OS was not different, aGVHD and cGVHD risk was greater and relapse risk was less with peripheral blood than with BM grafts.24 In another analysis in patients with AML in CR1, there were no differences in 2-year OS, 2-year LFS, cGVHD, relapse, or NRM, but grades III to IV aGVHD incidence was less with BM grafts than with PBSCT.51 In a small study by Bradstock et al, survival was significantly improved with PBSCT when compared with BM, but with significant differences between the cohorts; BM recipients received only 1 dose of PTCy, and PBSCT recipients received 2.50 In addition, CD34+ dose was doubled in patients receiving PBSCT compared with BM,50 which could have contributed to the inferior outcomes. When using BM grafts, higher nucleated cell graft dose has been associated with improved progression-free survival and OS in haplo-HSCT with PTCy.52 Thus, at centers with less experienced harvesters, there may be an advantage to using PBSCT, which typically has less potential for yielding low-dose grafts. Given the CIBMTR data showing less relapse with PBSCT,24 we typically choose PBSCT for patients with good functional status but high-risk hematologic malignancies, including those with minimal residual disease (MRD).
Do DSA matter?
Presence of circulating anti-HLA DSA in the recipient before transplantation increases the risk of primary graft failure.53 The incidence of DSA is highest in parous women, occurring in 52% (vs 31% in nulliparous females and 11% in males), but DSA can also be elicited in patients with high transfusion burden.14 The titer of DSA also influences outcomes, with mean fluorescence intensity (MFI) >2000 being associated with graft dysfunction,54 MFI >500015 being associated with graft failure, and MFI >10 000 being associated with very high incidence of graft failure.54 In the study by Ciurea et al, MFI >1500 was associated 25% engraftment compared with 95% for patients without DSA53 ; however, other data suggest that MFI <300055 or <5000 to HLA-A, HLA-B, and HLA-DR does not affect engraftment.56 In our current practice, we do not attempt desensitization if the recipient has anti–donor HLA antibodies of sufficient strength to result in a positive complement-dependent cytotoxicity assay result (associated with DSA on phenotype panels with MFI >10 000).14 Although what constitutes a prohibitive DSA level is unclear, we generally consider antibody levels to be weak to low with phenotype panel MFI values from 1000 to 3000, moderate from 3000 to 5000, and strong when >5000. Moderate to strong DSA levels are more frequently directed against familial haploidentical donors rather than MMUDs,14 which makes exploring MMUDs particularly important in cases of high DSA levels. Importantly, MFI values can differ between laboratories, and each institution should define their own MFI thresholds for graft failure risk.
Our desensitization process consists of tacrolimus and MMF starting 2 weeks before conditioning with every-other-day therapeutic plasma exchange (TPE) with post-TPE/IV immunoglobulin (IVIG). The number of TPE/IVIG treatments is influenced by the strength of DSA and cross-matching, being 3 to 4 for weak to moderately positive and 5 to 6 for patients with a strongly positive flow cross-match or a cross-match positive because of the presence of class II antibodies. Under that protocol, 15 patients underwent desensitization for high DSA. Fourteen patients achieved DSA levels below that consistent with a positive flow cross-match, all of whom engrafted after HSCT.57 Other desensitization platforms have been similarly successful. For instance, Ciurea et al employed a desensitization technique that included administration of an irradiated “buffy coat” prepared from 1 unit of blood and administered to the recipient on the day before transplantation as a method to sop up the DSA.15 However, this method has not been approved by the U.S. Food and Drug Administration, and therefore its use is not currently available outside of a clinical trial.15,58 Regardless of the platform, the goal of desensitizatoin is to reduce DSA to MFI <3000 on phenotype panels, to achieve a negative flow cross-match,56 or to become C1q testing negative through the clearance of complement-binding antibodies.15 Presence and strength of DSA are the most important aspects of haploidentical donor selection, with our practice being to choose the donor with the least DSA above all other factors and, if necessary, to use a MMUD59 rather than a haploidentical related donor for patients with prohibitive levels of DSA to relatives.
Do MRD and active disease matter?
Many studies in HLA-matched HSCT have shown the negative impact of the presence of MRD in the pretransplantation BM. Not surprisingly, pretransplantation MRD was also a risk factor for relapse after haplo-HSCT with PTCy, with a 2-year relapse incidence of 37% compared with 16% in patients who were MRD-negative before transplant.60 In a study of patients with active AML, patients who underwent haplo-HSCT, MUD-HSCT, or 9/10 HLA-MUD-HSCT were compared, and there was no significant difference in OS, LFS, relapse, or NRM between the groups.61 In all, the 2-year LFS was 25% in patients with active AML at the time of HSCT.61 Whether additional therapy to achieve MRD negativity before HSCT improves outcomes or whether it is a function of disease biology that cannot be overcome with further treatment remains to be seen. As such, for patients with MRD in whom additional pretransplantation therapy is not pursued, we prefer allografting with PBSCTs and/or for early cessation of immunosuppression, which was associated with less relapse in a small study of BM allografts.62 In addition, a clinical trial using maintenance therapies (eg, enasidenib, ivosidenib, idelalisib, gilteritinib, blinatumomab, venetoclax, and APR-246) or prophylactic posttransplantation immunotherapy (donor lymphocyte or natural killer cell infusion) to prevent relapse should be considered in these high-risk patients.
Clinical case continued
The patient discussed above received 1 cycle of high-dose cytarabine consolidation that was uncomplicated. His pretransplant BM showed CR without MRD. Sixty days from the start of consolidation, he began conditioning consisting of fludarabine, cyclophosphamide, and TBI followed by haploidentical BM graft with a total nucleated cell count of 2.9 × 107 total nucleated cells per kilogram from his brother, the donor. Postgrafting GVHD prophylaxis included PTCy on days 3 and 4, followed on day 5 by initiation of MMF and tacrolimus. He tolerated the procedure well, with his main complication being high fevers with hemodynamic stability from the time of graft infusion until after completion of PTCy on day 4, which was attributed to cytokine release syndrome (CRS) secondary to the HLA mismatches between donor and recipient. He had nausea and diarrhea during the first month. Neutrophil engraftment occurred on day 15, with platelet recovery occurring on day 19. The MMF was discontinued on day 35, and the patient’s gastrointestinal symptoms resolved shortly thereafter. He developed grade 1 overall acute GVHD after tacrolimus was discontinued on day 180 that improved with topical steroids. Days 90 and 180 BM biopsies showed remission with normal cytogenetics and 100% donor engraftment in both CD3+ and CD33+ cells.
Questions continued
How common is CRS after haploidentical graft infusion, and how is it treated?
Noninfectious fevers occur in 80% to 90% of haplo-PBSCT recipients between days 0 and 6 after transplantation. They typically resolve soon after completion of PTCy on day 4 and often do not require administration of steroids.63,64 These early fevers are associated with class II mismatching and higher CD3+ graft cell dose.65 Although the highest incidence of early fever has been demonstrated in haplo-PBSCT, haplo-BM allografts have also been associated with a higher incidence than HLA-matched BM allografts (13% after MAC MSD, 23% after MAC MUD, 44% after RIC haploidentical, and 84% after MAC haploidentical), but with these early fevers having no effect on survival.65 In contrast to BM grafts, haplo-PBSCT has also been associated with CRS, with 87% of early febrile patients meeting criteria, 12% of whom experienced grade 3 or 4 CRS.66 Transplantation-related mortality also rose in patients with grade 3 or 4 CRS, but symptoms could be alleviated with administration of tocilizumab.66 In the absence of severe CRS, diagnostic and supportive measures that include cultures and antipyretics are employed, and because of the difficulty of distinguishing sepsis and CRS in real time, broad-spectrum antibiotics are routinely administered. If grade 3 or 4 CRS develops before administration of PTCy, we would recommend administering tocilizumab if available and corticosteroids if tocilizumab is unavailable. Routine administration of corticosteroids before PTCy is generally avoided because it could theoretically decrease the efficacy of PTCy by preventing proliferation of alloreactive T cells, which leads them to be susceptible to PTCy and comprises one of PTCy’s several mechanisms of GVHD prevention.67 The management of CRS after haplo-HSCT is still a work in progress, with our suggestions merely reflecting our current clinical practice.
How long do we continue postgrafting immunosuppression?
In the original haplo-BMT PTCy study, MMF was administered 3 times daily for 35 days, and tacrolimus was administered to maintain a level of 5 to 10 μg/L through day 180 and stopped without a taper.1,42 However, a prolonged duration of postgrafting immunosuppression has the potential to increase infectious complications and calcineurin inhibitor–associated side effects and to impede graft-versus-leukemia responses. Emerging data in the HLA-matched setting suggest that total immunosuppression burden after PTCy may be less than with other strategies.68 As such, we are currently exploring in clinical trials whether we can reduce the duration of tacrolimus after haplo-HSCT with PTCy. In a recent publication, after haplo-BM allografting with PTCy, tacrolimus could be stopped as early as day 60 after transplantation.62 Clinical trials are ongoing in patients who have undergone allografting using haplo-PBSCT with PTCy to examine whether immunosuppression can safely be discontinued on day 90.
Clinical case continued
At 8 months after HSCT, our patient had 100% donor chimerism in the peripheral blood and BM, but he developed isolated testicular relapse that was treated with surgery, radiation, and prophylactic intrathecal chemotherapy. At 13 months after HSCT, he developed systemic relapse, with BM biopsy revealing 63% myeloblasts with 49% recipient DNA. Karyotype analysis at the time of relapse showed a complex karyotype. A small-nucleotide polymorphism array was performed on the BM and revealed a clonal 38.2-MB region of copy neutral loss of heterozygosity on the short arm of chromosome 6. The region of copy neutral loss of heterozygosity included the region where the recipient and donor haplotypes differed. Further molecular studies confirmed HLA loss.17 The patient underwent salvage chemotherapy with mitoxantrone, etoposide, and cytarabine and achieved a second CR with MRD negativity by flow cytometry. During salvage chemotherapy, we inquired regarding the availability of second-degree relatives. The patient had 5 nieces and nephews. We typed the children of the recipient’s haploidentical sister because these nieces and nephews also have a 50% chance of being haploidentical to the recipient. Importantly, the sister was HLA-disparate from the recipient’s original haploidentical brother donor, but she had medical problems preclusive of donation. Two of the recipient’s nieces were found to be haploidentical. The patient underwent a second fludarabine/cyclophosphamide/TBI haplo-PBSCT PTCy using a niece with a haplotype mismatch distinct from the original donor and received MMF and sirolimus prophylaxis. He experienced stage III aGVHD of the skin only, overall grade II aGVHD, that required systemic steroids started on day 65 posttransplant. Steroids were successfully tapered after 3 months, and sirolimus was stopped 1 month after steroid discontinuation. He was alive and in remission at last follow-up, approximately 14 months after HSCT.
Questions continued
How do we treat relapse after haplo-HSCT?
Leukemia relapse represents the most common cause of treatment failure and death in patients after HSCT, regardless of donor source. The approach for patients who relapse after haplo-HSCT is challenging, and there are no clear guidelines. Similar to HLA-matched HSCT, relapses occurring <6 months from HSCT are associated with poor outcomes,6 whereas later relapses can be successfully treated with additional chemotherapy followed by donor lymphocyte infusion (DLI), clinical trial, or second allogeneic transplantation. DLI is capable of inducing sustained remissions in relapse after haplo-HSCT, with a 31.3% CR rate in this disease.69 The incidences of grades II to IV and grades III to IV aGVHD after DLI are 20% and 15%, respectively, in the absence of GVHD prophylaxis.69 Patients with overt leukemia should receive reinduction chemotherapy, whereas in cases of low disease burden, it is reasonable to try hypomethylating agents before DLI. We suggest a starting dose of DLI of 106 CD3+ T cells per kilogram with escalation in nonresponders who do not develop GVHD to a maximum of 107 CD3+ T cells per kilogram.69,70
With the development of less toxic conditioning regimens and acceptable NRM, second allogeneic HSCTs have become a feasible option for patients experiencing disease relapse after a first HSCT. In the HLA-matched setting, several retrospective studies have shown that “medically fit” patients who relapse ≥6 months after the first HSCT may benefit from a second HSCT and achieve long-term disease-free survival that is at least proportional to the experience with DLI. Thus, a second haplo-HSCT from a relative who is HLA-mismatched to the original donor (and thus to the retained HLA) is a reasonable choice. Our early clinical data suggest that second HSCT is associated with a 4-year OS of 40%; however, longer survival was demonstrated when the second donor had a distinct haplotype mismatch from the initial HSCT donor.71 Our practice is to try DLI first, especially for early relapses, which could potentially be related to insufficient graft dose or incomplete immune reconstitution and reserve second HSCT for cases for DLI failure. However, we avoid DLI altogether and pursue a second HSCT if at the time of relapse there is no significant CD3+ donor chimerism or there is suspected HLA loss (discussed in more detail below).
What are the unique aspects of relapse after haplo-HSCT?
New insights into the biology of relapse have demonstrated that genomic HLA loss and downregulation of HLA are mechanisms frequently employed by leukemic cells to evade immune control (Figure 2).16,17 Loss of expression of the mismatched HLA haplotype has been described to occur in as many as 33% of patients with relapsed AML after haplo-HSCT, but it has also been demonstrated in myelodysplastic syndrome and myelofibrosis relapses.16-18 DLI is not anticipated to be effective in treating HLA loss relapse, but it still carries a risk of GVHD. Thus, in the case of confirmed HLA loss or HLA downregulation, DLI should be avoided. The same applies for performing a second HSCT from the original stem cell donor. We are currently exploring in patients who relapse after HSCT whether an HLA-mismatched second donor might actually be a better choice. When using a different haplotype-mismatched donor, the second donor’s T cells would be alloreactive to the mismatched HLA molecules retained on the leukemic blasts (Figure 2). Besides genomic loss of mismatched HLA alleles, downregulation of HLA class II molecules and upregulation of inhibitory T-cell ligands are likely important mechanisms of posttransplant relapse after haplo-HSCT.72 More work needs to be done to better understand the biology underlying graft-versus-tumor effects and posttransplant relapse and represents the next frontier. This work should also be complemented with a deeper understanding of how PTCy modulates alloreactivity and immune reconstitution, which represents an insight essential to the safe and effective integration of the growing immunological armamentarium, including cellular therapies, into haplo-HSCT.
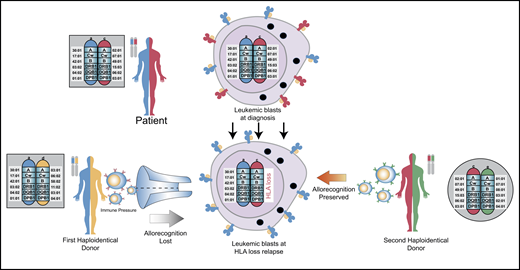
Rationale for using an alternative donor for second allogeneic transplantation. The patient’s 2 HLA haplotypes are shown in blue and red. The first donor shares a common blue haplotype with the recipient and a distinct yellow haplotype. At relapse, the leukemic blasts lose the mismatched red haplotype, which results in loss of cell surface expression of that mismatched HLA molecule. After relapse and subsequent chemotherapy to induce a remission, a second haploidentical donor is selected because they share the red haplotype with the patient, but lack the blue haplotype. This will allow the second donor’s immune system to recognize these disparate HLA molecules on the leukemic blasts to potentially elicit graft-versus-leukemic effects. Figure concept was influenced by two prior publications.8,73
Rationale for using an alternative donor for second allogeneic transplantation. The patient’s 2 HLA haplotypes are shown in blue and red. The first donor shares a common blue haplotype with the recipient and a distinct yellow haplotype. At relapse, the leukemic blasts lose the mismatched red haplotype, which results in loss of cell surface expression of that mismatched HLA molecule. After relapse and subsequent chemotherapy to induce a remission, a second haploidentical donor is selected because they share the red haplotype with the patient, but lack the blue haplotype. This will allow the second donor’s immune system to recognize these disparate HLA molecules on the leukemic blasts to potentially elicit graft-versus-leukemic effects. Figure concept was influenced by two prior publications.8,73
Conclusions
Haplo-HSCT with PTCy is an increasingly used platform, given its advantages of rapid identification of donors, low cost relative to other alternative donor strategies, simplicity of applying PTCy clinically, and comparability to HLA-matched HSCT. One of the most complex issues with haplo-HSCT is donor selection, given that multiple haploidentical donors are often available for a given recipient. No studies have prospectively compared first-degree relative haplo-HSCT with second-degree relative haplo-HSCT; however, the use of PTCy has made the latter approach appealing and safe and may make donor selection even more complex in the future. A significant barrier to haplo-HSCT is the high incidence of DSA in parous females, which can preclude familial haploidentical donors. Thus, clinical investigation of new desensitization platforms for patients with the highest levels of DSA (positive for either flow cross-match or complement dependent cytotoxicity) is warranted. In addition, in patients lacking a MUD, DSA are often lower to MMUD than to haploidentical relatives. Data supporting the safety of MMUD-BMT with PTCy suggest that this may be a viable alternative in this patient population. Finally, as with all HSCT, relapse remains the biggest barrier to successful haplo-HSCT and novel strategies to reduce relapse should be the focus of future investigation.
Acknowledgments
S.R.M. has received grant support from an American Cancer Society Institutional Research Grant. L.L. is supported by National Institutes of Health/National Cancer Institute grant 1P01CA225618.
Authorship
Contribution: L.L. and S.R.M. conceptualized and wrote the paper.
Conflict-of-interest disclosure: L.L. receives research support from Genentech and Merck, serves on a speaker’s bureau for Merck, serves as consultant and on advisory boards for AbbVie, and is a patent holder for WindMIL Therapeutics. S.R.M. declares no competing financial interests. Off-label drug use: None disclosed.
Correspondence: Leo Luznik, Johns Hopkins University, Cancer Research Building I, Room 2M88, 1650 Orleans St, Baltimore, MD 21231; e-mail: luznile@jhmi.edu.
This article was selected by the Blood and Hematology 2019 American Society of Hematology Education Program editors for concurrent submission to Blood and Hematology 2019. It is reprinted in Hematology Am Soc Hematol Educ Program. 2019;2019:513-521.
